Development of generic drug product by pharmaceutical industry is a scientific and technical approach which is totally different from developing a reference or innovator product. Most of the developing countries focus on developing the generic drug products because huge amount of investment is required for innovation and to develop reference product. The generic medicine has to be bioequivalent to the innovator drug and ensure the same biological effect with proper safety and efficacy. Nowadays, the pharmaceutical industries focus on the development of generic product as this does not require that much time and cost compared to the innovator company. But development of generic product is also difficult as it contains the same therapeutic efficacy as innovator. The development approach is based on the target market, i.e. US market, EU market. If a manufacturer targets the US market, then all excipients should be USP grade, analysis should be conducted by USP method or in-house method and stability studies as well. Prior and during the development of generic drug product API selection, dosage form selection, reference product selection and characterization, formulation development, analytical method development, tech transfer or submission batch are prime concern. Then again, bioequivalence study, drug registration procedure and commercialization of the generic product considering regulatory guidance of respective regulatory agencies and the approaches taken by the regulatory agencies for the development of registration of generic medicines are also crucial as well for the development of generic drug product. The aim of this study was to review the entire stage of a generic drug development by a generic pharmaceutical company.
1. Introduction
Developing a generic drug product in pharmaceutical industry is a scientific and technical approach which is totally different from developing an innovator product that takes several years and stages to be registered as an innovator product [1]. Generic products are sometimes termed as “Super Generic” or “Therapeutic hybrid” [2]. In terms of active ingredient(s), strength, dosage form, method of administration, quality, safety, performance characteristics, and therapeutic indication, bio-equivalence means that a generic drug product is virtually comparable to the originator product. Generic drug products are cost-effective than their respective brand products [3]. Pharmaceutical industries try to formulate and develop a generic product which is therapeutically equivalent to the innovator product. Though development of generic product does not require that much time and cost compared to the innovator company, but it is very difficult to develop a product containing the same therapeutic efficacy compared to innovator and to meet all the regulatory requirements as well [1].
2. Innovator and Generic Drug Products
Innovator products are also called “Reference Listed Drug” or “Patent Drug”. Innovator company or organization develop the medicinal product by trial and error and must have to conduct human trial or bio-availability study [4].
As mentioned earlier, the generic drug product has to be bioequivalent to the innovator product and ensure the same biological effect with proper safety and efficacy [4]. For example, “Zentiva Pharma UK Limited” invented an active, named “Dicycloverine Hydrochloride” and the marketed products was “Dicycloverine Hydrochloride 20 mg and 10 mg Tablets”. Another pharmaceutical industry “Teva UK Limited” developed bio-equivalent product by conducting bioequivalence study against the innovator product. So, the product of Zentiva is innovator and the product of Teva is generic product [5].
3. Basic Requirement of Generic Medicines
There are some mandatory rules to develop a generic product and the following parameters should be same as the innovator for developing any generic:
• The Active Pharmaceutical Ingredient and use.
• Dose and strength.
• The route of administration [4].
US FDA Criteria for a Generic Drug Product
United State Food and Drug Administration (US FDA) clarifies some basic requirement for a generic product. Some of them are as follow:
• Generic products are the pharmaceutical equivalent of innovator product where active ingredients are present in same proportion, in the same dosage form and given in the same route of administration.
• Generic product is identical regarding biological activity where potential bio-involution problems are not present.
• They are approved as safe, effective and have enough labels [4].
• They meet the same requirements for identity, strength, purity, and quality.
• They are made to the same exacting quality as innovator products, as defined by the FDA’s good manufacturing practice rules [2].
4. Development of Generic Product in Pharmaceutical Industries
The development of generic product in pharmaceutical industries requires lot of research and scientific review. To meet the target launching time, proper strategic planning is essential [1]. The Formulation scientist has to know and analyze the detail about the product to be developed and the target market as different market has different requirements. When a formulation scientist starts to develop a product for US, the product should comply to the physical and chemical parameter according to USP method and passes the bioequivalence study against the innovator product of US. The product developed according to US guidelines cannot be marketed in the EU/UK [6].
4.1. Development Strategies
Generic medication makers, on the other hand, must show that their formulation is bioequivalent to the brand-name equivalent in terms of quality and performance. When it comes to excipients, manufacturing processes, and performance qualities, an innovator drug product’s formulation development has few restrictions [7].
4.2. Patent Restriction Consideration
The listing of “relevant” patents and durations of “Exclusivity” for the approved drug product derives from the submission of an NDA with the FDA for a drug product developed with an NCE. The FDA’s “Approved Drug Products with Therapeutic Equivalence Evaluations,” sometimes known as the “Orange Book,” contains this listing. Before the expiration of the patent, innovator holds the permission to manufacturer for monopoly business. So before selecting the generic product to be developed the industries should consider the patent status and market share for sure [5].
4.3. Reference Product Selection and Characterization
As mentioned earlier, bio-equivalence of the generic product with reference listed drug (RLD) is the one of the prime criteria for development of generic product [8]. Reference product selection for US market can be easily done by literature search at US FDA database and can be easily identified by their NDA number [5]. In case of EU market, the formulation scientist has to review the “Public Assessment Report”, such as UKPAR or EPAR to identify the RLD. Again EMA, European Medicinal Agencies helps on regarding issues as well [9]. After having the RLD in pharmaceutical industries, the lab carries all the in-vitro tests, especially assay, dissolution of related substance. The prime target for a formulation scientist to review the dissolution data of reference drug and design the generic qualitative and quantitative composition to match with dissolution profile as % predictability can be determined through in-vitro–in-vivo correlation [10].
4.4. Target Market Selection
Target market selection determines and decides the way the company will develop the generic product, how to analyze the reference and generic product, the way stability test will be conducted, the way of selection of the batch size and so many variables. If a company targets the US market, then all excipients should be USP grade, analysis should be conducted by USP method or in-house method and stability studies as well. Again, if a bio-equivalence study is done against an EU reference product that is also acceptable for UK. In this case only dissolution profile comparison should be demonstrating between the EU reference product and UK reference product [1].
4.5. Manufacturing Feasibility
It is always important to assess the manufacturing feasibility in pharmaceutical industries. Feasibility report characterizes the whole process of the development of generic product. To register a generic product for US, EU or other countries, the manufacturer must have GMP or MHRA approved facility to manufacture the drug, to proceed stability test and for bulk manufacturing as well. Then the auditor will visit the plant as well before shipment and prior to dossier and other paper submission [11].
5. Formulation Development
Studies revealed, pharmaceutical industries have to bear about 14 – 18 percentage cost of their annual sales. Thus, developing generic product is a critical decision taken by the industry from where profit is uncertain considering the tight regulatory requirements [12].
5.1. Pre-Formulation Study
Before starting the lab-based formulation, extensive documentation and study is carried out to assess the API, e.g., appearance, solubility, salt form, pKa, assay, residual solvent, loss on drying, water content. Even bulk density and tapped density are analyzed. For specific molecule additional test may need to carry, e.g., “specific surface area” has to determine for “Nitrofurantoin Macrocrystal”. In preformulation study, drug-excipient compatibility has to be determined because it affect the final dosage form and stability of the drug and can modify the pharmacological effect as well “Pre-formulation” work must be reviewed prior to preparing actual trial formulations to achieve as much information related to the drug substance & reference product as possible [13].
5.2. Excipient Selection and Designing of Formulation Method
There are some basic requirements of pharmaceutical excipients:
• Chemically inert
• Cheap and available
• Exert the desired response for which they are used
From development point of view, nature of API, excipient used in reference product helps to design the formulation and selection of proper excipient. If any API is moisture sensitive, anhydrous lactose and cross povidone as filler and disintegrating agent will be right option rather than lactose monohydrate (filler) and maize starch (disintegrating agent). Formulation scientist has to assess such critical things during selection of proper excipient for formulation development [14].
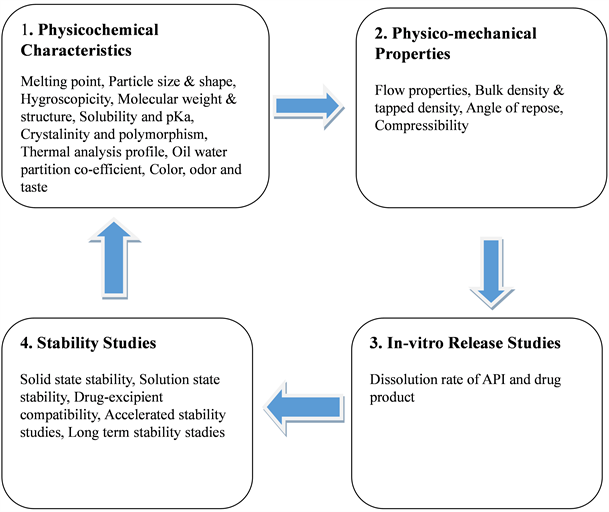
The following Figure 1 shows different parameters to be considered during preformulation studies [15].
Figure 1. Parameters to be considered during a Preformulation Study.
5.3. Selection of Manufacturing Method
Manufacturing method is the most critical decision for a formulation development, e.g., for solid dosage form manufacturing method can be Granulation or Direct compression. Again, granulation can be wet or dry. The method to be followed depends on the many things, like the characteristics of API; if API is heat and moisture sensitive, then wet granulation is not possible, similarly if particle size of API is larger, then the distribution of API may be ununiform in final products [6]. During the development a product by direct compression someone may find good physical and chemical result initially with small size batch. But in case of larger batch size, it may fail to comply uniformity of dosage unit by content. Then formulator may think about different approaches, like wet granulation if API is not heat and moisture sensitive and then product may comply with all specification. The scale-up plan must take into account the manufacturing technique used in smaller batch manufacturing, and the equipment employed in the development process frequently imposes many limits during scale-up operations. In the production of solid dosage forms, the following pharmaceutical unit procedures are used:
• Wet granulation
• Compression
• Extrusion
• Drying
• Dry blending
• Roller compaction
• Encapsulation
• Coating [6].
5.4. Lead Formula Identification
When pre-formulation, excipient selection and manufacturing method has been designed, then trial is planned to execute. The batch size should be as small as possible considering the expense of the industry. The prime objective is to match the dissolution profile with the reference. So, formulation scientist designs a unit formula composition considering the percentage of excipient. The formulation scientist will take suitable percentage of all excipient and will decide for target weight that allow for manufacturing [7].
Initially a small batch, e.g., 200 – 300 units are planned to manufacture and for that formula and method of manufacturing; steps in detail are documented and checked. Then materials are dispensed and manufactured up to final dosage form with proper documentation. Analytical method is also designed to carry on analysis simultaneously with formulation development. For first 2 – 3 small trial are done to just check the physical parameter like, weight variation, hardness, thickness, disintegration time and mainly to check the similarity with the dissolution profile with reference. If dissolution profile of test product matches with RLD and physical parameter are good enough, then formulator decides to scale up the formulation. The initial trial batches are called pilot batch or lab-based batch. The number of scales up batches may have to be taken depending upon the physical and chemical assessment of the finished products e.g., formulator brought a successful trial batch initially, but complicity may arise in the pilot batches. Then the formulation scientist has to rethink about the composition of the formulation and the method of manufacturing considering the complicity arises during the pilot batches. The formulation scientists may have to take new trial batch after taking multiple scale-up batches, even after a validation or optimization batch at GMP facility. The formula that passes the trial batch and scale up trial and passes to comply at least 3-month stability according to the ICH guideline can be called a lead formula. But it can be or need to change depending upon the situation arise as explained already [6].
Increase in batch size or scale-up are obtained by using larger, high-speed machinery that may necessitate co-relation to the process parameters determined using small scale equipment. Then the end goods must meet all predefined specifications, and the products from scaled-up batches must be physically and chemically identical, notably the dissolving profile [9]. Large scale batches or scale-up batches using modern technology has become essential in minimizing manufacturing costs in today’s competitive market [15]. The finished products must meet all predetermined specifications, and the products from scaled-up batches must be physically and chemically equivalent, specially the dissolution profile. “Informal stability” should be carried out under accelerated, long term and intermediate conditions as well for 2 – 3 months to ensure the stability of the product. But this stability data cannot be submitted as document, stability data can only be submitted from commercial site, not from R&D facility. The generic product is analyzed at monthly intervals mainly potency, dissolution, related substances. Stability of RLD should be carried as well for information only, mainly to assess the related substances [9].
This is important for the formulation scientist to validate and characterize the critical parameters and set specification during the initial formulation process. Critical parameters are as follow [6] :
Milling rate
Drying method
Rate of addition of the granulating vehicle
Screen size for initial sieving, during dry milling, during wet milling
Milling rates during wet granulation
Hardness of tablets
Screen-sizes
Shell size of capsule
Granulating time
Mixing order
LOD of granule
Blender type and blending time
Drying time
Lubrication time
Drying condition
Machine speed during compression and encapsulation
Speed of chopper and agitator during granulation
Feeder speed
The formulation scientist is responsible to manufacture of the exhibit batch. Batch can be manufactured as a sub-batch using similar equipment as the scale-up equipment and using the same raw materials. As for example, using a 30 kg capacity granulator can be used to manufacture 60 kg batch-size. In this case, granulation sub-lots would be required (30 × 2) to complete the batch manufacture. During each granulation, drying, milling, and blending operations need be optimized according to protocol. Manufacturing of tech transfer or submission batches should be carried out under GMP facility by maintaining the cGMP regulations. Samples from the executed batches must be tested to the laboratory under cGMP facility with validated method. Proper documentation needs to be prepared which describes the whole process, termed as Batch Manufacturing Record and for packaging, batch packaging record. Stability must be conducted as per requirements [6]. A proper and documented validation protocol ensures the above. A validation master plan should include the following:
Sampling plan
Critical process and product parameters
Critical process steps
Testing plan
Purpose of the study
Acceptance criteria
Personnel responsibilities
During the manufacturing process, extensive samples are collected and tested to validate the process the samples are tested to appropriate testing using validated analytical methods. The validation report should include the following items [6] :
Critical process steps studied
Attachments of copies of the executed batch records
Aim of the validation study
List of raw materials used in the manufacturing process
List of manufacturing equipment
Product and process acceptance criteria evaluation
Recommendations by the validation department
Data collected and their analysis
Statistical analysis of results
After careful evaluation of the validation documentation the manufacturing process is released for regular production.
5.5. Analytical Method Development
Analytical method development and validation is one of the major steps in the process of API selection for generic drug development, proper characterization of API, sensitivity precision, accuracy for quantification of the drug in a dosage form, linearity, and assurance of inactive ingredient compatibility [6].
Purity, identification, assay, dissolution profile, disintegration time, LOD, water content, and other analytical test methods are used to develop and validate data. A proper analytical approach ensures product quality and accelerates the development process by reducing development time [6]. The following parameters are used to qualify the validity of an analytical procedure:
Quality of the procedure itself
Accuracy
Precision
Specificity
Limit of Detection
Quantitation Limit
Robustness
Before the establishment of a test method, the laboratory personnel have to assess the following requirements:
Technical
Practical
Regulatory
Validation
Transfer
Time limitation [9].
Development of accurate methods is an essential element for the development of generic product which will ensure correct decisions being made from data for stability studies and formulation development as well. In order to assist other plant activities such as packaging development and cleaning validation, analytical development and validation must have a timeline when manufacturing a drug product. Depending on the situation and regulatory needs, a validated method can still be changed. It is required for describing the test method’s capabilities and supporting the validation report when it is audited. In a pre-approval examination, the regulatory agency giving a drug product approval will request this data [6].
5.6. Site Transfer Batch or Submission Batches
When formulation has been developed and analytical method has been transferred to the manufacturing site, the generic company planned to conduct their site transfer or submission batches. These batches will be included in the dossier and for submission. Sometimes an optimization batch is taken followed by two consecutive validation or submission batches. Sometimes multiple optimization batches need to conduct until a successful optimization batch is done. Sometimes depending upon the capacity of RMG batch size can be adjusted. If the product the coated, then size of the coating pan is a limiting factor to determine the batch size. This happens especially for low dose drug with low average weight. Batch size, stability protocol of the developed product depends on the market being targeted [9]. The following Table 1 shows requirement of different regulatory bodies [6].
Countries
Stability Condition
Stability Commitment while Filling
Number of Submission Batches
Bio equivalence Requirement
Dissolution Requirement
EU
Long term
Accelerated
Intermediate
On Three commercial batches
Two batches: one lacks units
Fast, Fed if required
Minimum in 3 media
US
One batch: one lacks units
Fast and Fed both
Only official media
Russia
Optional
Three batches: one lacks units
Fast and Fed both
Minimum in 3 media
Asia
Accelerated Intermediate
N/A
Three representative batches
Only for epileptic drug
N/A
Africa
Long term Accelerated
Optional
Two batches: one lacks units
Fast or Fed
Minimum in 3 media
Table 1. Comparative regulatory requirement for different countries.
The process validation department uses the signed Master Manufacturing Document to create process and cleaning assessment protocols. Generally, at least three batches of each strength of drug product are required for validation, and the protocols must monitor and manage all important steps in the production process [6].
The in-process blend, final blend, finished product should be tested by the validated method already discussed. Batch can be made as a sub-batch with the same scale-up equipment and raw materials as the scale-up batch. The drying, milling, and blending activities must all be optimized according to protocol during each granulation. Manufacturing of tech transfer or submission batches should be carried out under GMP facility by maintaining the cGMP regulations. Samples from the executed batches must be tested to the laboratory under cGMP facility with validated method. Proper documentation needs to be prepared which describes the whole process, termed as Batch Manufacturing Record and for packaging, batch packaging record. Stability must be conducted as per requirements [6]. A proper and documented validation protocol ensures the above. The following should be included in a validation master plan:
Sampling plan
Critical process steps
The study purposes
Responsibilities of personnel
Critical parameters of process and product
Testing plan
Criteria of acceptance
Extensive samples are gathered and evaluated during the manufacturing process to confirm the process. The samples are tested using proven analytical procedures. The following items should be included in the validation report:
Critical process steps studied
Aim of the validation study
List of manufacturing equipment
Recommendations by the validation department
Statistical analysis of results
Attachments of copies of the executed batch records
List of raw materials used in the manufacturing process
Product and process acceptance criteria evaluation
Data collected and their analysis.
The manufacturing method is released for regular production after a thorough examination of the validation paperwork [6].
5.7. Stability Protocol
The QC and QA division at the commercial location should carefully create the stability protocol based on the intended market. These data will be filed for dossier approval to the appropriate regulatory authorities. The protocol should include a stability study design like the below:
Package sizes
Sampling time points
Strengths
Bracketing
Depending on the demand, it should define the environmental parameters for accelerated and long-term stability, although it is safer to proceed stability at each condition. Based on the data of stability the following decisions are made:
Development of the Shelf-life
Shelf-life specifications during the expiration period
Assignment of Expiration Dates
Stability on an Annual Basis
Extending the Period of Expiration
Packaging
Storage Requirements [9].
6. Bioavailability and Bio-Equivalence
As mentioned earlier, Generic drug product has to be bioequivalent and interchangeable with the innovator or reference product. Bioequivalence study can be done in fasting or fed condition. Sometimes both studies are required depending on the regulatory bodies. The European medicine Agency (EMA) and FDA have released the guidance the guidance for the bioavailability and bioequivalence. FDA publishes a list approved products with their Equivalents in a book called “Orange Book” [16].
For solid dosage form and generic drug development, achieving the desired bioavailability is the major challenge. Then AUC between the test drug and reference drug should be more than 90% confidential interval. Bioavailability can be in lower range even though where similarity factor is more than 50 because bioavailability depends on various factors, like membrane permeability, human physiology, drug-food interaction, drug-drug interaction etc. That’s why, bioequivalence study is so unpredictable.
Bioequivalence is the evidence that a generic product is bioequivalent with the reference product. Bioequivalence is more critical in case of drug with narrow therapeutic index [16].
7. Fundamental Assumption and Bioequivalence Study Design
A generic drug product that has been approved can be used in place of the reference drug product. A bioequivalence research is frequently designed after employing a crossover design to establish comparison within individual participants. Bioequivalence may then be assessed using reliable statistical methods while taking into account the pre-specified regulatory requirements for bioequivalence. Bioequivalence evaluation is one of the most contentious problems, as bioequivalence does not always indicate therapeutic equivalent, and approval of generics does not ensure bioequivalence. Though bioequivalence assumption, is often difficult, but carried out pharmaceutical company by conducting of clinical trials. When evaluating bioequivalence for generic medication approval, there are four factors to consider.
1) Although the absorption characteristics of the drugs are not identical, they are therapeutically comparable.
2) Although the patterns of drug absorption are comparable, they are not therapeutically identical.
3) The absorption characteristics of generic and reference drugs are not the same, and they have different equivalency.
4) Despite the similarities in medication absorption patterns, they may not be therapeutically comparable [10].
EMA considers generic medication products and RLDs having the same active ingredient to be bioequivalent if the rate and extent of absorption are assessed using plasma drug concentration-time curves following administration in the same molar dose [3].
Conduct of Bioequivalence Trial
Bioequivalence is the evidence that a generic product is bioequivalent with the reference product which comprise a set of stage:
Subject Selection
Washout
Blood sampling
Statistical Methods.
After a successful bioequivalence study, the generic company started to work on dossier submission and filling of other regulatory documents for approval of generic product [10].
8. Regulatory Aspects for the Development of Generic Product
The regulatory standards of different nations throughout the world differ, making it extremely difficult to produce a single drug that can be presented for approval in all of them at the same time. Regulatory agencies ensure the safety, efficacy, and quality of all generic products, as well as the production and distribution processes. The regulatory authority’s primary aim is to ensure that pharmaceutical goods are developed in accordance with the country’s regulatory requirements, and generic companies aim to meet those requirements [9].
8.1. Regulatory Requirement for EU
The EU, which comprises of 27 member states, has one of the most well-regarded and well-organized regulatory systems in the world. Iceland, Liechtenstein, and Norway are also members of the European Free Trade Association.
EMA is responsible for the scientific evaluation of generic products developed by pharmaceutical industries which is applicable for human and medicine drugs. Prior to 2004, the European Agency for the Evaluation of Medicinal Products, or EMA, was known as the European Medicines Evaluation Agency [9].
8.2. Regulatory Requirement for USA
The abbreviated New Medicine Application is the application for registering a generic drug in the United States, which is one of the most demanding regulatory authorities in the world. The US FDA, like the EU, ensures that manufacturing, medication development, and testing are carried out in accordance with rules and that everything is documented as required. The ICH created a standardized structure for submitting applications for registering generic medication products, and the paper addresses generic drug registration requirements in the form of a dossier for market authorization in the United States [8].
9. Abbreviated New Drug Application
Abbreviated New Drug Application is the application submitted to the FDA for the approval of a generic drug product and for marketing authorization. Though innovator product has submitted human trial study, but the generic industries need not to submit any clinical and animal studies to prove their safety and efficacy as they will submit the bioequivalence study which is compared to the innovator drug. It has created opportunities for the generic pharmaceuticals for developing and marketing generics within 180 days, even though it took minimum 18 months for the final approval of ANDA by the FDA [8].
9.1. Types of Certifications
The generic companies have to prepare one of the four certifications for each patent:
1) Paragraph (I): The generic firm certifies to the FDA that no patent information on the reference name medication has been provided.
2) Paragraph (II): Generic company gives a certification that, the patent has expired already.
3) Paragraph (III): Generic company gives a certification that, the patent is not expired yet, but will be expired soon before their generic product enters into the market.
4) Paragraph (IV): Generic company gives a certification that, the patent information is invalid.
FDA will approve the ANDA of a generic company immediately when the generic makes a paragraph I or II certification and after the patent’s expiration date, FDA will approve a paragraph III certification any time. But approval of paragraph IV certification is little bit tricky [8].
9.2. Common Technical Document (CTD)
CTD is a standard technical document that makes it easier to submit a single generic drug registration application to many countries at the same time. There are five modules in CTD where point to be mentioned that, the content of module-1 is not same for all regulatory bodies. That’s why Module-1 is not a part of CTD. The following Table 2 shows the different modules and their respective contents [8].
9.3. Registration Requirements for Generic Drug Products in the US
The followings are mandatory for US submission and registration of generic product:
1) For drug approval applications, US FDA guideline papers and FDA sections (e.g., 505 (b) for NDA and 505 (j) for ANDA) must be followed in the compilation of the dossier.
2) For submission of the drug applications (NDA/ANDA), the eCTD is mandatory.
3) The applicant or an approved agent may submit the application to FDA directly.
4) The applications are different from each other:
For new drug—NDA
For generic drug—ANDA
For biological application—BLA.
1) Application information, financial certification, patent information, administrative information needs to be identical.
2) Package insert is mandatory.
3) Cartons and Proposed Labels should be similar to that of RLD.
Modules
Contents
1
Prescribing information and all the administrative information.
2
Clinical and non-clinical overview, pharmacology, pharmacokinetics, toxicology, overall quality summaries.
3
Complete pharmaceutical and technical aspects, pharmaceutical development report, Stability studies of the product, Packing and storage conditions
4
Complete pharmacological and toxicological study reports as non-clinical report
5
Clinical study Reports
Table 2. Different modules of CTD and their respective contents.
4) Clinical investigators and financial disclosure should be clearly stated.
5) In module 1, request for waiver of in-vivo BE studies need to be provided.
6) Environment Assessment Statement should be provided.
7) In terms of API and excipient, a statement should be supplied for residual solvents.
8) CEP is not applicable.
9) For expedited studies, three months of stability data is submitted at the time of submission.
10) In eCTD, the USFDA requires a study tagging file and structured product labeling [8].
9.4. ANDA Regulatory Review Process
Firstly, applicant submits an ANDA to the “Office Generic Drugs” or “Centre for Drug Evaluation and Research”. The document room staff then processes the ANDA by giving it an ANDA number and stamping a received date on the cover letter of the ANDA. Then ANDA then sent to a consumer safety technician who is responsible for reviewing the preliminary sections of the ANDA checklist. During ANDA filing, the drug’s bioequivalence, microbiology and chemistry, as well as the labeling, are all taken into account. The filing review is completed within 60 days. When proposal inspection is reviewed and other documents are reviewed and found satisfactory, then ANDA is approved [8].
10. Bio Equivalence Review Process
Both the innovator and generic medication should have the same pharmacological properties, such as
Same strength
Same dosage form
Same route of administration
They should be equivalent when assessed under same condition
The area under the curve (AUC) and the highest concentration of the drug (Cmax) are used to determine bioequivalence. A generic product is considered bioequivalent to an innovator product if the 90 percent confidence interval for the mean AUC and relative mean Cmax is 80 percent to 125 percent [8].
11. Labeling Review Process
The review procedure guarantees that the labeling of both generic and innovator drugs is consistent. After the final level administrative review and individual disciplines have resolved their shortcomings, and if there are any unexpired patents or exclusivities given to the RLD, the application will either get a complete approval or a preliminary approval letter [8].
12. Conclusion
Nowadays, most of the pharmaceutical countries focus on developing the generic drug products rather than concentrating on the innovation. This is more applicable in developing countries. One of the reasons behind is the amount of investment required for innovation and to develop reference product. Then development and registration of generic drug product considering regulatory aspect requires a long-term procedure as most of the Government health authorities of the different countries demand product development history, manufacturing direction, composition declaration, analytical test methods, quality related product specifications, stability report, test method validation reports, GMP certificate, product registration certificate from parent country, quality management certificate, cGMP certificate of API as well as manufacturing facility, Bioequivalence study report, DMF, CEP for EU/UK market to ensure quality and safety of drug product depends not only on generic firm but also on bulk API production company. Therefore, every pharmaceutical industry should be responsible for the development of generic drug product considering the regulatory aspects and safety of the drug product as well.
Conflicts of Interest
The authors declare no conflicts of interest regarding the publication of this paper.
References
Handoo, S., Arora, V., Khera, D., Nandi, P.K. and Sahu, S.K. (2012) A Comprehensive Study on Regulatory Requirements for Development and Filing of Generic Drugs Globally. International Journal of Pharmaceutical Investigation, 2, 99-105.
Barei, F., Pen, C.L. and Simoens, P.S. (2013) The Generic Pharmaceutical Industry: Moving beyond Incremental Innovation towards Re-Innovation. Generics and Biosimilar Initiative Journal, 2, 13-19.
Ho, K. and Mcdonald, K. (2012) Development and Manufacture of Drug Substances-Chemical and Biotechnological/Biological Entities. Generic and Biosimilar Initiative Journal, 1, 142-144.
Nikam, N.R., Vakhariya, R.R. and Magdum, C.S. (2019) Generic vs. Brand Medicines: An Overview. Asian Journal of Pharmaceutical Research, 9, 109-115.
Chawla, K., Tofighi, T., Agarwal, A., Thomas, J. and Mondald, T. (2014) A Global Comparison between Brand-Name and Generic drugs. Indian Journal of Pharmacy Practice, 7, 23-28.
Pramod, K., Tahir, M.A., Charoo, N.A., Shahid, H., Ansari, S.H. and Ali, J. (2016) Pharmaceutical Product Development: A Quality by Design Approach. International Journal of Pharmaceutical Investigation, 6, 129-138.
Terblanche, N.S. (2008) New Pharmaceutical Product Development: Barriers to Overcome and Opportunities to Exploit. Journal of Commercial Biotechnology, 14, 201-212.
Rafi, N., Sandeep, D.S. and Narayanan, A. (2018) Regulatory Requirements and Registration Procedure for Generic Drugs in USA. Indian Journal of Pharmaceutical Education and Research, 52, 544-549.
Sravika, S., Bhavana, R., Sharmila, V., Anusha, S., Mounica, N.V.N. and Nagabhushanam, M.V. (2017) A Comprehensive Study on Regulatory Requirements for Development and Filing of Generic Drugs Globally. Journal of Pharmaceutical Innovation, 6, 153-158.
Chow, S.C. (2015) Bioavailability and Bioequivalence in Drug Development. WIREs Computational Molecular Science, 6, 304-312.
Rahi, S. and Rana, A. (2019) Role of ICH Guideline in Registration of Pharmaceutical Products. International Journal of Drug Regulatory Affairs, 7, 14-27.
Yousefi, N., Mehralian, G., Rasekh, H.R. and Yousefi, M. (2017) New Product Development in the Pharmaceutical Industry: Evidence from a Generic Market. Iranian Journal of Pharmaceutical Research, 16, 834-846.
Bachhav, A.A., Ahire, S.A. and Jadhav, A.G. (2019) Preformulation Study of Piroxicam. International Journal of Pharmaceutical Sciences and Research, 10, 811-818.
Chaudhari, S.P. and Patil, P.S. (2012) Pharmaceutical Excipient: An Overview. International Journal of Advances in Pharmacy, Biology and Chemistry, 1, 21-32.
Chaurasia, G. (2016) A Review on Pharmaceutical Preformulation Studies in Formulation and Development of New Drug Molecules. International Journal of Pharmaceutical Sciences and Research, 7, 2313-2320.
Muntha, P. (2015) Bioavailability & Bioequivalence Studies—Pharmaceutical Importance. Research and Reviews, 4, 55-58.








(2) Pathway: Uncovering Drug Development Trends and Regulatory Requirements")










![Fig 2. Sales for 8 medication-device combination products in 100-top sellers in the US from 2011–2013 [22]](https://www.drugpatentwatch.com/blog/wp-content/uploads/2019/03/Fig-2.-Sales-for-8-medication-device-combination-products-in-100-top-sellers-in-the-US-from-2011–2013-22.png "Is Patent “Evergreening” Restricting Access to Medicine/Device Combination Products?")




