Executive Summary: Navigating the Global Regulatory Maze
The journey of bringing a new drug or medical device to market is a monumental undertaking, fraught with immense financial investment, protracted timelines, and a high probability of failure. For pharmaceutical and medical technology companies, understanding the intricate regulatory landscapes of the United States, primarily governed by the Food and Drug Administration (FDA), and the European Union, overseen by the European Medicines Agency (EMA) for drugs and the Medical Device Regulation (MDR) and In Vitro Diagnostic Medical Device Regulation (IVDR) for devices, is not merely a compliance exercise; it is a strategic imperative. This report delves into the core distinctions in approval processes, timelines, costs, and underlying philosophical approaches between these two major global markets. By dissecting these nuances, businesses can transform regulatory knowledge into a powerful competitive advantage, optimizing R&D investments, streamlining market entry, and ensuring long-term product success.
Introduction: The Strategic Imperative of Regulatory Acumen in Pharma and MedTech
The High Stakes of Drug and Device Development
The path from a groundbreaking scientific discovery to a commercially available drug or medical device is notoriously challenging. It is a marathon, not a sprint, demanding relentless innovation, rigorous testing, and substantial capital. Consider the pharmaceutical sector: the average cost of developing a new prescription drug hovers around an astonishing $2.6 billion, with the entire process, from initial discovery to market approval, typically spanning 10 to 15 years . This staggering figure accounts not only for successful compounds but also for the vast majority that never reach patients. Indeed, only about 12% of drugs that enter human clinical trials eventually receive FDA approval, and a mere 1 in 10,000 compounds that begin in preclinical research ultimately make it to market . These statistics paint a stark picture of the inherent risk and financial commitment involved.
The medical device industry, while often characterized by shorter development cycles than pharmaceuticals, faces its own formidable challenges. Bringing a new medical device to market can take anywhere from 3 to 7 years. The financial outlay is considerable, with costs potentially reaching upwards of $30 million for Class II devices cleared via the 510(k) pathway, and soaring to $94 million for high-risk Class III devices requiring Premarket Approval (PMA). A significant portion of these costs, approximately $24 million for Class II devices, is specifically allocated to FDA-required activities like clinical studies. The sheer scale of these investments underscores why every strategic decision, particularly those concerning regulatory pathways, carries profound financial and operational consequences. For companies navigating these complex waters, a deep understanding of regulatory requirements and philosophies is not just beneficial; it is absolutely essential for survival and prosperity.
Report Scope: Unpacking US (FDA) and European (EMA, MDR, IVDR) Approval Landscapes
This report embarks on a comprehensive exploration of the intricate regulatory landscapes governing drugs and medical devices in two of the world’s most influential markets: the United States and the European Union. In the US, the Food and Drug Administration (FDA) stands as the singular, centralized authority. Across the Atlantic, the European Medicines Agency (EMA) takes the lead for centralized drug approvals, while medical devices and in vitro diagnostics are governed by the robust Medical Device Regulation (MDR) and In Vitro Diagnostic Medical Device Regulation (IVDR), respectively, with a crucial role played by independent Notified Bodies.
The objective is to conduct a rigorous comparative analysis, dissecting not only the procedural similarities that facilitate global development but, more importantly, the profound differences that shape development pathways, market access strategies, and ongoing post-market obligations. By illuminating these distinctions, this report aims to equip business professionals—from R&D leaders and regulatory affairs specialists to market access strategists and investors—with the knowledge to transform regulatory compliance from a perceived hurdle into a powerful lever for competitive advantage.
The U.S. Regulatory Framework: FDA’s Centralized Authority
The FDA operates as a single, centralized governmental authority, overseeing the entire drug and device development process across the United States . This centralized structure offers inherent advantages in terms of common rules and a potentially unified decision-making process, though it also means a single point of stringent review for all products entering the market.
Decoding the FDA Drug Approval Process
The FDA drug approval process is a meticulously structured, multi-stage journey designed to ensure the safety and efficacy of new pharmaceutical products before they are made available for sale in the U.S. .
Discovery, Preclinical Studies, and the IND Gateway
The arduous journey of drug development commences in the laboratory, where researchers engage in the discovery and identification of promising drug compounds . This foundational phase, often spanning 3 to 6 years and costing between $300 million and $600 million, focuses on a deep understanding of the drug’s pharmacological activity, its toxicity profile, bioavailability (how much of the drug is absorbed), and pharmacokinetics (how the drug moves through the body) . These critical assessments are conducted through a combination of in vitro (test tube or cell culture) and in vivo (animal) studies . Key studies during this period include toxicology studies to identify adverse effects, as well as in vitro and in vivo tests to determine safety and biological activity. Researchers also examine absorption, distribution, metabolism, and excretion (ADME) to ensure the drug reaches its intended target and is properly processed by the body . Furthermore, chemistry tests are performed to establish the drug’s purity, stability, and shelf life, alongside manufacturing tests to determine large-scale production feasibility and optimal dosing, packaging, and formulation.
These preclinical studies are not simply scientific exercises; they are subject to stringent FDA regulations known as Good Laboratory Practices (GLP) for Nonclinical Laboratory Studies (21 CFR 58) . Compliance with GLP ensures that the data generated are credible, reproducible, and acceptable for regulatory review, covering areas such as personnel, facilities, equipment, and operations . The meticulous results from this preclinical research form the bedrock for the next critical step: the submission of an Investigational New Drug (IND) Application .
The IND application is a formal request submitted to the FDA, seeking permission to initiate human clinical trials . This comprehensive application must include all preclinical data, details on the drug’s composition, manufacturing information, the proposed clinical trial protocols, and information about the investigators involved. The FDA has a 30-day window to review the IND application. If no clinical hold is issued within this period, the sponsor is granted permission to proceed with human trials. It is crucial to understand that IND approval is merely a “green light” for clinical research; it is not a final marketing approval. Its primary purpose is to ensure that the preclinical data have been adequately evaluated and that human subjects will not be exposed to unreasonable risks during the subsequent clinical investigation.
Clinical Trials: The Phased Journey to Efficacy and Safety (Phases I-III)
Once the IND is approved by the FDA, the investigational drug embarks on its clinical testing journey, typically progressing through three major phases of human trials . This structured, sequential approach is designed to systematically de-risk drug candidates, progressively gathering more comprehensive safety and efficacy data, though this methodical process inherently contributes to the lengthy overall development timelines.
Phase I: Safety and Dosage This initial phase involves a small group of participants, typically 20 to 100 healthy volunteers or, in some cases, patients with the target condition . The primary objectives are to assess the drug’s safety profile, its tolerability, and its pharmacokinetics (how the drug is absorbed, distributed, metabolized, and excreted in the human body) . Researchers also examine how the experimental drug interacts with the body and how the body processes it. This phase usually spans several months. If the results indicate serious adverse events, the FDA may not permit progression to Phase II, often leading to the discontinuation of the treatment’s development.
Phase II: Efficacy and Side Effects Following successful Phase I completion, the drug moves to Phase II, which involves a larger cohort of 100 to 300 patients who actually have the target condition . The central focus shifts to determining the optimal dose range and establishing initial efficacy of the drug in treating the disease . Safety continues to be rigorously monitored. Trial sponsors often assign participants to different treatment groups, some receiving varying doses, while a control group might receive the current standard of care or a placebo. This phase typically takes several months to two years to complete. Failure to demonstrate efficacy superior to placebo or the occurrence of unexpected serious adverse events can lead to the FDA withholding permission to proceed to Phase III, often resulting in the termination of the drug’s development.
Phase III: Large-Scale Efficacy and Monitoring The final and most extensive phase of clinical testing, Phase III, involves a much larger group of patients, ranging from 1,000 to 3,000 individuals with the target disease . The primary goals are to confirm the drug’s effectiveness on a larger scale, monitor for less common side effects that might only appear in larger populations, and compare the new treatment’s performance against existing treatments . This phase can last from 1 to 4 years. The expansion of the group size is crucial to ensure that the results are statistically significant and broadly applicable to the general population affected by the target disease or condition. If the treatment does not demonstrate superior efficacy to placebo or standard of care, or if it causes unexpected serious adverse events, the FDA may not grant permission to submit a New Drug Application (NDA). The structured, sequential nature of these clinical trials, with clear progression criteria between phases, aims to systematically de-risk drug candidates. This rigorous, step-by-step evaluation, while paramount for patient safety and generating robust data, also significantly contributes to the lengthy development timelines, as evidenced by the decade-plus average from discovery to market. The “gatekeeping” function of the FDA at the conclusion of each phase, where progression can be halted due to safety or efficacy concerns, ensures a high standard of review but also means substantial time and investment can be lost if a drug fails in later stages.
New Drug Application (NDA) Submission and Rigorous Review
Upon successful completion of all three clinical trial phases, the pharmaceutical company compiles all its findings into a comprehensive New Drug Application (NDA) . The NDA is a formal request to the FDA to approve a new pharmaceutical drug for sale in the United States . This extensive dossier serves three core purposes: to confirm the drug’s safety and efficacy for its intended use, to establish that its benefits outweigh its risks when used as labeled, and to verify that the drug can be manufactured with high quality and consistency according to Good Manufacturing Practices (GMP) .
The NDA submission is the culmination of years of drug development, preclinical studies, and clinical trials. It must include a full report of clinical trials, manufacturing information, proposed labeling, a thorough risk-benefit analysis, and safety updates . Often, companies hold a pre-NDA meeting with the FDA to clarify regulatory requirements, confirm labeling expectations, and address any potential gaps in clinical data before the formal submission . A well-structured NDA, typically formatted according to the Common Technical Document (CTD) format, is essential for a smooth FDA review process. Key components include a cover letter summarizing the submission, the official Form FDA 356h, and detailed information on intellectual property protections and regulatory exclusivity claims.
Once submitted, the FDA initiates its rigorous review process. The agency first takes 60 days for an initial filing review to determine if the application is complete and acceptable . Following this, the review period typically lasts 10 months for a standard review or a shortened 6 months for a priority review . During this intensive period, FDA review teams meticulously examine all submitted clinical and statistical data, conduct pre-approval inspections (PAI) of manufacturing facilities to ensure compliance with GMP and quality control processes, and thoroughly evaluate the proposed labeling and promotional materials provided by the applicant company . The FDA may also convene advisory committees, comprised of independent scientists, clinicians, and regulatory experts, to review the NDA data, evaluate the drug’s benefit-risk profile, and vote on a recommendation for approval, rejection, or additional studies. After the comprehensive review, the FDA issues either an approval letter, allowing marketing, or a complete response letter, detailing deficiencies that require resolution before approval can be granted.
Post-Marketing Surveillance: The Ongoing Vigilance of Phase IV
FDA approval of a new drug is not the final chapter in its regulatory journey; rather, it marks the beginning of an ongoing commitment to patient safety through post-marketing surveillance, commonly referred to as Phase IV studies . This continuous monitoring is a critical component of the drug’s lifecycle, extending long after the product enters the market.
In this crucial phase, the focus shifts to observing the drug’s performance in real-world scenarios, which often differ significantly from the controlled environment of clinical trials. Phase IV studies aim to identify long-term safety concerns, uncover rare side effects that might not have appeared in smaller pre-market populations, investigate drug interactions with other medications, and evaluate the drug’s use in special populations, such as pregnant women, children, or elderly patients, who may have been excluded or underrepresented in earlier trials .
The FDA possesses statutory and regulatory authority to require manufacturers to conduct post-market safety studies and clinical trials. This is particularly pertinent for therapies approved via expedited pathways like Accelerated Approval, where initial data might rely on surrogate endpoints and uncertainties may remain at the time of pre-market authorization . Similarly, post-market studies may be required for deferred pediatric studies or products approved under the Animal Efficacy Rule. These requirements ensure that any potential serious risks associated with the drugs are thoroughly assessed.
Manufacturers are obligated to submit annual reports detailing the status of these post-market requirements (PMRs) and post-market commitments (PMCs). Beyond formal studies, post-market surveillance can leverage various real-world data sources, including patient registries and natural history studies, which collect observational data over time. The FDA also champions initiatives like Sentinel, an active surveillance program designed to efficiently monitor drug safety on a massive scale by analyzing healthcare data. This robust post-market surveillance framework, including mandatory Phase IV studies and the Sentinel system, reflects a profound commitment to continuous safety monitoring, especially for drugs that benefited from expedited approval pathways. This approach acknowledges that while speed to market can be vital for unmet medical needs, it must be balanced by ongoing vigilance to ensure long-term patient well-being.
Expedited Pathways: Accelerating Access to Critical Therapies
In response to urgent public health needs and to facilitate faster access to innovative treatments, the FDA has developed and implemented several expedited approval programs . These programs are designed to accelerate the development and review processes for drugs that treat serious or life-threatening conditions or address unmet medical needs, without compromising the agency’s standards for quality, safety, and efficacy .
The four primary Expedited Review Programs are:
Fast Track Designation: This program is granted to drugs that treat serious conditions and have the potential to address an unmet medical need . A key benefit of Fast Track is increased opportunities for interaction between the drug sponsor and FDA reviewers throughout the drug development and review process. This early and frequent communication can help resolve questions and issues more quickly, potentially leading to faster approval. An additional feature is “rolling review,” which allows a drug sponsor to submit completed sections of their New Drug Application (NDA) or Biologics License Application (BLA) for review even before the entire application is compiled, further accelerating the overall process .
Breakthrough Therapy Designation (BTD): Reserved for drugs that treat a serious or life-threatening condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on a clinically significant endpoint . BTD fosters intensive FDA guidance and organizational commitment, encouraging close collaboration and efficient development.
Priority Review (PR): This designation signifies that the FDA’s goal is to take action on a drug sponsor’s marketing application within six months, significantly shortening the standard review time of 10 months . To be eligible, a drug must treat a serious condition and, if approved, provide a significant improvement in safety or effectiveness over existing treatments. Priority Review Vouchers, awarded for developing treatments for neglected tropical diseases, rare pediatric diseases, or medical countermeasures, can also be redeemed or sold to obtain this expedited review.
Accelerated Approval (AA): This pathway allows for earlier approval of drugs for serious conditions that fill an unmet medical need, based on a “surrogate endpoint” or “intermediate clinical endpoint” that is reasonably likely to predict a clinical benefit . Surrogate endpoints, such as a decrease in biomarker levels or improved imaging results, can be measured earlier than primary outcomes like survival, thereby accelerating market access. However, drugs approved via Accelerated Approval often come with rigorous post-authorization study commitments to confirm the clinical benefit .
The FDA’s aggressive use of these expedited pathways, particularly Accelerated Approval, implicitly shifts some of the evidence-gathering burden to the post-market phase. While this strategy is crucial for accelerating patient access to potentially life-saving therapies for serious conditions, it necessitates robust post-market surveillance commitments from manufacturers to confirm long-term safety and effectiveness. This calculated regulatory risk-benefit decision prioritizes timely access, balanced by mandated continued data collection once the drug is on the market.
Navigating the FDA Medical Device Approval Process
The FDA’s approach to regulating medical devices is fundamentally risk-based, meaning that the potential threat a device poses to patient safety directly dictates the level of regulatory scrutiny it must undergo for market entry .
Risk-Based Classification: Class I, II, and III Devices
Medical devices in the United States are categorized into three distinct classes, each corresponding to a different level of risk and, consequently, different regulatory requirements . This clear, risk-based classification system allows for a more efficient pathway for lower-risk devices while reserving the most rigorous oversight for high-risk, life-sustaining technologies.
Class I (Low-Risk Devices): These devices present minimal potential harm to the user and are often simpler in design . Examples include elastic bandages, manual stethoscopes, and enema kits . The majority of Class I devices (around 95%) are exempt from premarket notification (510(k)) requirements and are primarily subject only to “General Controls” . General Controls include basic requirements such as establishment registration, device listing, proper labeling, adherence to Quality System Regulations (QSRs), and recordkeeping. Approximately 47% of all medical devices fall into this category .
Class II (Moderate-Risk Devices): Devices in this class are those for which General Controls alone are deemed insufficient to provide a reasonable assurance of safety and effectiveness. Therefore, they require additional “Special Controls” to ensure their safety and efficacy. These Special Controls can include performance standards, post-market surveillance, patient registries, and specific labeling requirements. Examples of Class II devices include powered wheelchairs, infusion pumps, and CT scanners . A significant majority of Class II devices (43% of all medical devices) must undergo the 510(k) premarket notification process .
Class III (High-Risk Devices): These devices represent the highest risk category. They are typically those that sustain or support human life, are implanted within the body, or present a potential unreasonable risk of illness or injury . Examples include pacemakers, orthopedic implants, artificial heart valves, and breast implants . Due to their critical nature and potential for harm, Class III devices are subject to the most stringent regulatory requirements, primarily the Premarket Approval (PMA) process . Only about 10% of medical devices fall into this high-risk category. The FDA’s explicit link between device class and required controls (General, Special, PMA) demonstrates a tiered regulatory approach. This means manufacturers of Class I devices face significantly lower hurdles compared to Class III devices requiring extensive PMA, allowing for more efficient market access for less complex innovations while ensuring critical oversight for higher-risk products.
Premarket Pathways: 510(k) Clearance, De Novo Classification, and PMA Approval
The FDA employs distinct premarket pathways for medical devices, tailored to their classification and whether a similar device is already on the market.
510(k) Premarket Notification: This is the most commonly used submission pathway, required for many Class II devices and some Class I products . To achieve clearance, a manufacturer must demonstrate “substantial equivalence” (SE) to a legally marketed predicate device . Substantial equivalence means the new device has the same intended use and the same technological characteristics as the predicate, or, if it has different technological characteristics, that these differences do not raise new questions of safety or effectiveness and the device is at least as safe and effective as the predicate . The submission package includes device classification, a detailed device description, a comparison with the predicate device(s), intended use, proposed labeling, and applicable performance testing data . The FDA aims to reach a decision on 95% of 510(k) submissions within 90 FDA days .
De Novo Classification Request: This pathway was specifically designed for novel devices that are low-to-moderate risk but lack a legally marketed predicate device . Without the De Novo pathway, such devices would automatically be classified as Class III, requiring the more rigorous PMA process . A successful De Novo request allows the FDA to create a new device type and classification regulation, typically classifying the device as Class I or Class II . This is a crucial innovation enabler, as the newly classified device can then serve as a predicate for future 510(k) submissions of similar devices, making it easier for other manufacturers to bring comparable products to market . De Novo submissions must include a thorough risk assessment, clinical and nonclinical testing data, a benefit-risk justification, and proposed Special Controls . The FDA encourages manufacturers to submit a pre-submission for a De Novo request to receive early feedback on eligibility and required testing. This pathway bridges the gap between truly novel, lower-risk devices and the stringent PMA process, fostering a more dynamic MedTech landscape in the US.
Premarket Approval (PMA): This is the most rigorous and demanding pathway, mandatory for Class III devices . The PMA process requires extensive scientific evidence, including well-controlled clinical studies conducted under an Investigational Device Exemption (IDE). The application must include detailed design history, biocompatibility and sterilization validation, risk analysis, comprehensive labeling review, and robust manufacturing controls. PMAs are also subject to review by an FDA Advisory Panel, composed of external experts, and carry comprehensive post-approval requirements. An approved PMA is, in essence, a private license granted to the applicant for marketing that specific medical device.
Investigational Device Exemption (IDE): Paving the Way for Clinical Studies
Before a medical device that poses a significant risk can be tested in human subjects, or when an approved device is being investigated for a new indication, an Investigational Device Exemption (IDE) from the FDA is typically required . The IDE allows an unapproved device to be shipped and used in a clinical study to collect safety and effectiveness data, which will ultimately support a marketing application (e.g., a PMA).
Devices considered “significant risk” (SR) include implantable devices such as cardiac pacemakers, orthopedic implants, and stents, as well as certain non-implantable devices like computer software used for prenatal risk evaluation. For such devices, both an FDA IDE and Institutional Review Board (IRB) approval are mandatory before clinical investigation can begin. The FDA typically reviews an IDE application within 30 days.
However, not all investigational device studies require an FDA IDE. For devices deemed “non-significant risk” (NSR), an FDA IDE is not needed if the IRB agrees with the sponsor’s determination that the device poses non-significant risk . Examples of NSR devices include daily wear contact lenses, conventional endoscopes, and externally worn monitors for insulin reactions. The IRB plays a crucial role in assessing risk for these studies and ensuring participant protection . The IDE submission process ensures that human subjects are protected from unreasonable risks and that the clinical investigation is scientifically sound, providing the necessary data to support future marketing applications.
Quality System Regulation (QSR) 21 CFR Part 820: The Backbone of Device Quality
The FDA’s Quality System Regulation (QSR), codified in 21 CFR Part 820, serves as the cornerstone for ensuring the safety and quality of medical devices throughout their lifecycle in the United States . This regulation outlines the current Good Manufacturing Practice (cGMP) requirements that medical device manufacturers must meticulously follow.
The scope of 21 CFR Part 820 is broad, encompassing “the design, manufacture, packaging, labeling, storage, installation, and servicing of all finished devices intended for human use,” including the facilities and designs utilized for these processes . It mandates the establishment and maintenance of a robust Quality Management System (QMS) that ensures products are safe, effective, and audit-ready.
Key components and requirements of the QSR include:
Management Responsibility: This subpart covers the establishment of a quality policy, allocation of resources, and planning for quality, ensuring management’s commitment to quality and its implementation .
Quality System Requirements: Details the organizational structure, responsibilities, and authority within the QMS, including provisions for internal quality audits and personnel training and experience .
Design Controls: A critical section outlining controls for the entire design and development process, from planning and input to output, review, verification, validation, and transfer to production. Proper implementation of design controls is essential for generating the Design History File (DHF) and Device Master Record (DMR).
Document Controls: Specifies requirements for document approval, distribution, and management of changes, ensuring that all documentation is controlled, available, and accurately reflects the current state of processes and products.
Production and Process Controls: Mandates the development and monitoring of production processes to manufacture devices according to specifications, including instructions, Standard Operating Procedures (SOPs), environmental controls, and equipment maintenance.
Record Keeping and Reporting: Emphasizes accurate and comprehensive record-keeping for all activities, from raw data to protocols and final reports, which must be stored securely and made available for FDA inspection. Manufacturers must maintain a Device Master Record (DMR) that includes device specifications, production process specifications, quality assurance procedures, and packaging/labeling specifications.
Compliance with QSR is not merely a pre-market hurdle; it is a continuous obligation. The FDA conducts regular inspections to ensure ongoing adherence to 21 CFR Part 820, utilizing techniques like the Quality System Inspection Technique (QSIT). Violations can result in significant consequences, including Form 483 observations and warning letters. Adhering to these guidelines is crucial because it ensures that the data generated are credible, reproducible, and acceptable for regulatory review, underpinning the safety and effectiveness of devices on the market.
Post-Market Surveillance for Medical Devices
FDA approval or clearance for a medical device is not the endpoint of regulatory oversight; rather, it signifies the transition to continuous post-market surveillance. This ongoing monitoring ensures the device’s safety and effectiveness once it is available for public use.
While all medical devices are subject to general post-market surveillance activities, the intensity and specific requirements can vary significantly based on the device’s classification and premarket pathway. Devices approved through the Premarket Approval (PMA) pathway, typically Class III high-risk devices, are subject to greater user fees over their lifespan and more stringent change control requirements compared to devices cleared via 510(k) or De Novo pathways. This reflects the FDA’s commitment to continuous vigilance for the highest-risk products.
For devices, particularly those approved through expedited programs like the Breakthrough Devices Program (BDP), the FDA explicitly leverages post-market data collection to address any uncertainties that may have existed with pre-market data. This approach is an integral part of the FDA’s benefit-risk determination, allowing for earlier patient access to life-saving devices while ensuring that safety and effectiveness are continuously monitored in real-world settings. The FDA can require manufacturers to conduct specific post-market studies for certain devices, especially when an unexpected serious risk is identified or to monitor biologically plausible safety issues. This robust post-market oversight ensures that even with expedited market access, patient safety and device effectiveness are maintained throughout the product’s lifecycle.
This strong emphasis on post-market oversight, balanced against the speed of approval, particularly for 510(k) and De Novo pathways, demonstrates a core philosophy: the FDA aims for “reasonable assurance” of safety and effectiveness at the time of approval, complemented by continuous real-world performance monitoring. This approach allows for quicker market entry for many devices while maintaining a vigilant eye on their long-term impact.
The European Regulatory Framework: EMA and the Evolving EU Landscape
The European Union’s regulatory landscape for drugs and medical devices is characterized by a multi-stakeholder approach, involving the European Medicines Agency (EMA) for centralized drug approvals, and a system of independent Notified Bodies for medical devices under the Medical Device Regulation (MDR) and In Vitro Diagnostic Medical Device Regulation (IVDR), alongside national competent authorities . This contrasts with the FDA’s single, centralized authority.
The EMA Drug Approval Process: Centralized and Decentralized Routes
The EMA plays a pivotal role in the authorization and monitoring of human medicines across the European Union . Unlike the FDA’s single national process, the EU offers several distinct routes for drug approval, providing flexibility but also introducing layers of complexity .
Preclinical Development and Clinical Trial Application (CTA)
Similar to the United States, the development of a new drug in the EU commences with an extensive preclinical phase. During this stage, comprehensive studies are conducted in laboratory settings and on animal models to rigorously assess the drug’s safety, efficacy, and potential side effects before any human exposure . These preclinical investigations must strictly adhere to Good Laboratory Practice (GLP) guidelines to ensure the reliability and integrity of the generated data. The EMA actively supports developers during this early phase, offering scientific advice and guidance on appropriate tests and studies . This includes “protocol assistance,” a specialized form of scientific advice specifically reserved for medicines designated as “orphan drugs” (those for rare conditions).
Once a sponsor has gathered sufficient preclinical data to support human studies and believes the potential benefits of the therapeutic outweigh any known safety issues, they submit a Clinical Trial Application (CTA) to the relevant European regulatory authorities . This application serves as a formal request for permission to proceed with clinical trials in human subjects. If the regulatory agency deems the data satisfactory and the proposed human studies ethically sound, permission is granted, allowing the investigational drug to advance into the clinical development phase. This early dialogue and scientific advice from the EMA are designed to facilitate the preparation of robust applications and ensure a smooth validation and assessment procedure later in the development process.
Clinical Trials: European Phases I-III
European clinical trials generally mirror the phased structure observed in the US, following the internationally recognized guidelines established by the International Council for Harmonisation (ICH) . This adherence to ICH guidelines is a key factor that can influence development strategies for companies seeking multi-regional approval, as it often allows for a single, well-designed global trial program to potentially satisfy both US and EU regulatory bodies.
Phase I: These trials involve a small number of healthy volunteers or, in certain cases, patients with the targeted condition . The primary objectives are to confirm that the medicine behaves as expected from laboratory tests, assess its safety and tolerability in humans, and understand its pharmacokinetics (how the drug is absorbed, distributed, metabolized, and excreted) .
Phase II: Expanding on Phase I, these trials involve a relatively small number of patients who have the specific condition the drug is intended to treat . The aim is to gather more detailed information about the optimal doses to use and to confirm initial indications of the drug’s efficacy . Safety monitoring continues throughout this phase.
Phase III: This phase involves a much larger group of volunteers, similar to US trials, to examine the safety and efficacy of the experimental treatment on a broader scale. The goal is to ensure that the results are statistically robust and applicable to the general population affected by the target disease or condition. During this phase, researchers meticulously document and report any side effects experienced by patients, often requiring long-term exposure to the drug for proper assessment.
While the fundamental phases of clinical trials are largely harmonized between the US and EU, the EMA’s emphasis on ICH guidelines and its willingness to permit global clinical trials that meet EU regulatory standards can significantly influence development strategies for companies. This approach offers an efficiency advantage for multinational pharmaceutical companies, potentially reducing the need for redundant trials in different regions and streamlining the overall clinical development timeline.
Marketing Authorisation Application (MAA) and Procedural Nuances
In the European Union, the formal request for market approval of a new drug is known as a Marketing Authorisation Application (MAA) . Unlike the FDA’s single, centralized approval pathway, the EU offers a multi-pathway system for drug authorization, providing strategic flexibility but also introducing a layer of complexity for manufacturers .
Centralised Authorisation Procedure: This is the primary route for innovative medicines and is compulsory for certain categories of products, such as those derived from biotechnology (e.g., genetic engineering), advanced therapy medicines (like gene or somatic cell therapy), and medicines for specific diseases (e.g., HIV/AIDS, cancer, diabetes, neurodegenerative diseases, and orphan medicines) . It is also optional for other new active substances or those representing a significant therapeutic, scientific, or technical innovation, or whose authorization would be in the interest of public health at the EU level. Under this procedure, pharmaceutical companies submit a single MAA directly to the EMA. The EMA’s Committee for Medicinal Products for Human Use (CHMP) conducts a comprehensive scientific assessment of the application and provides a recommendation on whether the medicine should be marketed . Based on the CHMP’s positive opinion, the European Commission then takes a legally binding decision to grant the marketing authorization, which is valid in all EU Member States and European Economic Area (EEA) countries (Iceland, Liechtenstein, and Norway) . The core assessment timeline for the EMA is 210 calendar days . However, this timeline is subject to “clock stops” during which the applicant prepares responses to EMA’s questions, and also includes the European Commission’s decision, which is issued within 67 days of receiving EMA’s recommendation . Consequently, the centralized procedure can typically extend to 14 months, or even around 16 months if an oral explanation is required from the applicant.
National Authorisation Procedures: These procedures are used to obtain marketing authorization in a single EU Member State . This route is suitable if the drug is intended for a specific country or if the applicant does not initially wish to market the product across the entire EU . Most national competent authorities aim to review and grant the MA within 210 days, though specific timelines can vary by Member State, as individual countries maintain their own submission requirements within the general EU framework. Many older medicines and most generic medicines or those available without a prescription were authorized at the national level.
Decentralised Procedure (DCP): This procedure is designed for products that have not yet been authorized in any EU Member State and allows for simultaneous authorization in several chosen EU countries . The applicant selects a Reference Member State (RMS) to lead the assessment, and all involved Member States must reach a unanimous decision on whether to approve the application. The DCP typically follows a 210-day timeline, with structured clock-stop periods for applicant responses.
Mutual Recognition Procedure (MRP): The MRP is utilized when an applicant already possesses a marketing authorization in one EU Member State (the Reference Member State) and seeks to have this authorization recognized in other EU countries (Concerned Member States) . Similar to the DCP, the RMS leads the assessment, and the procedure follows fixed timelines, typically around 90 days after the dossier is submitted to the Concerned Member States.
The EU’s multi-pathway system for drug approval offers considerable flexibility, allowing manufacturers to tailor their market entry strategy based on the nature of their product and their commercial objectives. However, this flexibility inherently introduces complexity, requiring a nuanced understanding of each pathway’s eligibility criteria, timelines, and procedural requirements. This strategic choice is paramount for optimizing market access and resource allocation within the diverse European market.
Post-Authorisation Pharmacovigilance and Risk Management Plans (RMPs)
EMA authorization is not the final regulatory step; rather, it marks the beginning of continuous, rigorous post-authorisation pharmacovigilance. Once a medicine has been authorized for use in the EU, the EMA and individual EU Member States constantly monitor its safety, taking swift action if new information suggests the medicine is no longer as safe and effective as initially thought .
The EMA has a dedicated committee, the Pharmacovigilance Risk Assessment Committee (PRAC), specifically responsible for assessing and monitoring the safety of medicines on the market. This committee ensures that the EMA and Member States can respond quickly to detected safety issues, leading to necessary actions such as amending product information, restricting use, or even suspending a medicine to protect patients. The EMA can also initiate a review of a medicine or class of medicines upon request from a Member State or the European Commission, often triggered by concerns related to a medicine’s safety, the effectiveness of risk minimization measures, or its overall benefit-risk balance.
A key distinction in the EU’s approach to post-market safety is the mandatory requirement for manufacturers to submit a Risk Management Plan (RMP) for all new medicinal products, even if no additional risk minimization measures are initially deemed necessary. The RMP is a comprehensive document that outlines how identified and potential risks associated with the medicine will be managed throughout its entire lifecycle. This contrasts with the FDA’s Risk Evaluation and Mitigation Strategies (REMS), which apply only to specific medicinal products with serious safety concerns already identified.
The RMP typically includes routine risk minimization tools such as the Summary of Product Characteristics (SmPC) for healthcare professionals and the Patient Information Leaflet (PIL). However, for medicines with specific serious risks, additional risk minimization activities may be required. These can include educational programs for healthcare professionals, patients, and caregivers, “Dear Healthcare Professional Communications” (DHPCs), pregnancy prevention programs, controlled access programs, or controlled distribution systems.
The EMA’s mandatory RMP for all new medicinal products, in contrast to the FDA’s targeted REMS, reflects a more comprehensive, proactive, and integrated approach to risk management from the outset of a drug’s market life. This implies a fundamental philosophical difference: the EMA embeds risk management as an inherent, continuous process for every new drug, proactively planning for potential risks, whereas the FDA’s REMS are often more reactive or targeted to specific high-risk scenarios. This broader, upfront approach by the EMA aims for a more holistic safety profile throughout the product lifecycle.
Understanding EU Medical Device and IVD Regulations (MDR & IVDR)
The European Union’s regulatory framework for medical devices underwent a profound transformation with the implementation of the Medical Device Regulation (MDR) (Regulation (EU) 2017/745), which became fully effective in May 2021, and the In Vitro Diagnostic Medical Device Regulation (IVDR) (Regulation (EU) 2017/746), effective in May 2022 . These regulations replaced older directives, introducing significantly stricter requirements for clinical data, post-market surveillance, and the pivotal role of independent Notified Bodies .
EU Device Classification: MDR (I, IIa, IIb, III) and IVDR (A, B, C, D)
The EU’s classification systems for medical devices and in vitro diagnostic (IVD) medical devices are designed to categorize products based on their intended use and potential risk of harm to users and public health. This classification dictates the conformity assessment route and the level of regulatory scrutiny required. The EU MDR categorizes medical devices into four classes, while the IVDR assigns IVD devices to four risk classes, with increasing levels of risk requiring more stringent oversight .
MDR Medical Device Classes:
Class I (Lowest Risk): These devices pose a minimal danger to patients and are primarily used for non-invasive procedures and basic functions . Examples include hospital beds, corrective glasses, and thermometers . This class is further subdivided:
Class Is: Indicates a sterile device (e.g., personal protection kits).
Class Im: Indicates a device with a measuring feature (e.g., thermometers).
Class Ir: Comprehends a reusable surgical instrument (e.g., surgical instruments). Most Class I devices, except for Is, Im, and Ir, can be self-certified by the manufacturer, without the mandatory involvement of a Notified Body .
Class IIa (Moderate Risk): These devices present a moderate risk potential, higher than Class I but lower than Class IIb and Class III. They are typically surgically invasive devices intended for transient (less than 60 minutes) or short-term (between 60 minutes and 30 days) use. Active medical devices intended for diagnosis and monitoring are also often classified as Class IIa. Examples include catheters, hearing aids, and surgical clamps .
Class IIb (Medium/High Risk): Class IIb medical devices have a greater impact on patient health than lower classes and are often implantable or long-term surgically invasive devices (continuous use for more than 30 days). Examples include contact lenses, surgical lasers, ventilators, blood bags, and intensive care monitoring equipment .
Class III (Highest Risk): These devices represent the highest risk category, sustaining or supporting life, or being implanted, with significant potential for illness or injury . Examples include pacemakers, orthopedic implants, prosthetic heart valves, defibrillators, and aneurysm clips . These devices are subject to the most stringent regulatory oversight.
The EU MDR utilizes 22 classification rules, grouped into categories like non-invasive, invasive, active, and special rules, which assess the device’s intended use, materials, and duration of use .
IVDR In Vitro Diagnostic (IVD) Device Classes:
The EU IVDR categorizes IVD medical devices into four risk classes (A, B, C, and D) based on their intended use and potential risk to individual and public health, with Class D being the highest risk .
Class A (Low Individual Risk, Low Public Health Risk): Includes devices with minimal risk to individuals and public health. Examples are general laboratory equipment, specimen receptacles, and buffers . Notified Body involvement is generally not required, except if the device is sterile.
Class B (Moderate Individual Risk, Low Public Health Risk): These devices carry moderate risk to the individual but limited public health implications. Examples include pregnancy tests and cholesterol tests . These devices require Notified Body certification.
Class C (High Individual Risk, Moderate Public Health Risk): Devices in this class pose a serious individual risk and moderate public health implications. Examples include genetic testing and cancer diagnosis IVDs .
Class D (Highest Risk): Class D devices represent the highest risk category, posing a high individual and a high public health risk. They are typically used for screening, diagnosing, or monitoring life-threatening and highly transmissible infectious diseases, such as HIV or Hepatitis C . Given their critical public health role, any diagnostic error could lead to widespread consequences, and they undergo the most stringent regulatory oversight.
The EU MDR/IVDR classification system is notably more granular and, in many cases, more stringent than the FDA’s, particularly for devices that might be considered lower risk in the US. For instance, specific devices like joint prostheses are classified differently in the US and the EU. This increased granularity, coupled with the fact that some Class I devices in the US are even exempt from QSR compliance , implies that devices might often be categorized into a higher risk class in the EU. A higher classification under MDR/IVDR directly translates to a greater need for Notified Body involvement and more extensive clinical data requirements , thereby increasing the regulatory burden and associated costs for manufacturers seeking market entry in Europe.
The Pivotal Role of Notified Bodies and CE Marking
For the majority of medical devices and in vitro diagnostic (IVD) medical devices in the European Union, obtaining a certificate from an independent third-party “Notified Body” is an indispensable step to affix the CE mark . This CE mark is a mandatory conformity marking that signifies a product’s compliance with EU health, safety, and environmental protection standards, and it is required for the distribution and sale of such medical devices within the EU market .
Notified Bodies (NBs) are independent certification institutions that are designated and overseen by the Competent Authority of an EU Member State. Once recognized by one Competent Authority, they are acknowledged by all other competent authorities to perform conformity assessments for products across the entire EU. Their primary responsibility is to provide conformity assessment services according to the EU MDR and IVDR, taking into account all relevant guidance documents and harmonized standards.
The core tasks performed by NBs include:
Certifying and auditing Quality Management Systems (QMS): NBs conduct thorough audits of manufacturers’ QMS to ensure they meet the stringent requirements of the MDR and IVDR.
Assessment of technical documentation: They meticulously review the technical documentation submitted by manufacturers, which includes design history, risk management files, and clinical evaluation reports.
Verification of products through sampling and testing: For certain device classes, NBs may conduct or oversee sampling and testing to verify product conformity.
The new MDR and IVDR have introduced significant obligations for Notified Bodies, and they are currently undergoing a substantial revamp to comply with these increased responsibilities. This includes a projected increase in the number of IVDs that will require Notified Body assessment for the first time—an estimated 35,000 IVDs. This system, which relies on a network of independent third-party assessors, stands in stark contrast to the FDA’s centralized review process in the United States.
The reliance on Notified Bodies for CE marking introduces a decentralized element to EU device approval. While this system aims to facilitate market access across diverse member states, it can lead to variability in interpretation and capacity issues. The fact that “different Notified Bodies may have slightly different interpretations of the regulation” suggests a potential for inconsistency in the assessment process. Furthermore, concerns exist regarding “insufficient availability and expertise in the Notified Body system” under the new MDR and IVDR. This distributed responsibility, while a strength in accommodating diverse national contexts, can paradoxically lead to longer approval timelines and greater uncertainty for manufacturers compared to the FDA’s single, centralized review process.
Clinical Evaluation Report (CER): The Cornerstone of EU Device Approval
Under the stringent requirements of the European Union Medical Device Regulation (MDR), medical device manufacturers are mandated to conduct a thorough clinical evaluation to demonstrate conformity with the General Safety and Performance Requirements (GSPRs) outlined in Annex I of the MDR . The results of this evaluation are meticulously documented in a Clinical Evaluation Report (CER) . This is a fundamental requirement for all medical devices, including Class I devices, if any clinical claims are made regarding their performance or safety. The CER is not a one-time submission; it must be continuously updated throughout the device’s entire lifecycle to reflect new clinical data and findings from post-market surveillance .
The process of preparing a CER is systematic and rigorous, typically involving several key stages:
Clinical Evaluation Plan (CEP): The manufacturer must first establish a comprehensive Clinical Evaluation Plan (CEP). This document outlines the objectives, methodology, and timeline for the clinical evaluation, specifying the types of clinical data to be used and the criteria for assessing the device’s safety and performance .
Identification and Appraisal of Data: This stage involves identifying all available relevant clinical data related to the device under evaluation. This includes pre-market clinical study data, post-market clinical follow-up (PMCF) study data, sales data, and post-market surveillance data generated or held by the manufacturer . Data from scientific literature, including both favorable and unfavorable findings, are also systematically searched and qualitatively appraised to assess the level of evidence, potential bias, and limitations .
State of the Art Review: A critical component of the CER is the “state of the art” review. This section describes the current standard of care, alternative treatments, and existing medical practices relevant to the device and its intended purpose . The goal is to specify objective parameters for safety and performance, derived from aggregate data sources like meta-analyses and society guidelines, which will then be used to determine the acceptability of the subject device’s benefit-risk ratio. A systematic literature search process is paramount for reproducibility and to minimize bias in this review.
Analysis of Clinical Data: Once the data is gathered, manufacturers must analyze it comprehensively to assess the device’s safety and performance, comparing it against the intended use, risk profile, and applicable regulatory standards.
Benefit/Risk Analysis: The collected and analyzed data are then used to perform a thorough benefit/risk analysis. The benefits of the device are summarized and weighed against its identified risks. The risks identified in the clinical evaluation must align with the manufacturer’s risk management documents, instructions for use, and promotional materials . The acceptability of the benefit-risk profile is determined by comparing the device’s performance against the parameters established in the state of the art review.
The MDR places a much greater emphasis on collecting or generating stronger clinical evidence than its predecessor, the Medical Device Directive (MDD) . For implantable and Class III devices, the MDR specifically mandates that clinical data from high-quality sources must include data obtained with the device under evaluation itself. This heightened emphasis on the Clinical Evaluation Report (CER) and robust clinical evidence, even for lower-risk devices, signifies a fundamental shift towards a more “drug-like” evidence standard for devices in Europe. This convergence in the type of evidence expected, even if the approval pathways differ, directly impacts development costs and timelines for device manufacturers, as it necessitates more extensive and continuous data generation.
Post-Market Surveillance (PMS) and Post-Market Clinical Follow-up (PMCF)
Under the European Union Medical Device Regulation (MDR), post-market surveillance (PMS) is not merely an optional activity but a comprehensive and mandatory system integrated into the manufacturer’s Quality Management System (QMS) . Its purpose is to proactively and systematically gather experience from the use of devices once they are on the market, continuously monitoring their performance and safety throughout their entire lifetime .
The PMS system for each device must be described in a detailed PMS plan and includes several key activities:
Assessment of Feedback: Manufacturers are required to document and evaluate all incoming feedback about their device, distinguishing between serious and non-serious incidents.
Trend Analysis: The MDR mandates documenting all non-serious incidents and conducting statistical analysis to identify trends, aiming to detect potential incidents or dangerous situations before they escalate .
Databases and Registries: Manufacturers must screen relevant databases, incident report databases (like SOUP), and technical, specialist, and regulatory literature to identify new risks, off-label uses, and updates to applicable standards or common specifications.
Communication: The PMS system defines methods for communication with competent authorities, Notified Bodies, authorized representatives, importers, distributors, users, and patients.
A crucial component of the EU’s post-market framework is Post-Market Clinical Follow-up (PMCF). PMCF is a continuous process designed to update the clinical evaluation of a device by proactively collecting and evaluating clinical data from the use of CE-marked devices in humans throughout their expected lifetime. PMCF is mandatory for all device classes under the MDR, including legacy devices (those approved under previous directives).
The PMCF plan is an integral part of the overall post-market surveillance system and is reviewed by the Notified Body as a binding commitment during the CE-certification process. The objectives of PMCF are comprehensive:
To confirm the safety and performance of the device throughout its expected lifetime.
To identify previously unknown side effects and monitor identified side effects and contraindications.
To identify and analyze emergent risks based on factual evidence.
To ensure the continued acceptability of the device’s benefit-risk ratio.
PMCF activities can involve general methods, such as gathering clinical experience, collecting feedback from users, and screening scientific literature . More specific methods may include dedicated PMCF studies or the evaluation of suitable patient registries. Failure to develop and execute a comprehensive PMCF plan can lead to significant consequences, including major non-conformances and potential regulatory sanctions. For devices that gained certification through equivalence, PMCF is particularly essential to ensure that clinical data is collected for the actual device in use.
The EU MDR’s stringent and continuous PMCF requirements, mandatory for all device classes, signify a proactive and long-term commitment to real-world evidence. This approach aims to lead to earlier identification of safety issues and emergent risks , potentially enhancing patient safety across the Union. However, it also imposes significant ongoing compliance costs and burdens on manufacturers, requiring robust QMS integration and continuous data collection, analysis, and reporting even after market entry .
A Comparative Lens: Unveiling Key Differences and Strategic Implications
Understanding the nuances between the US and EU regulatory systems is paramount for businesses seeking global market access and a sustainable competitive advantage. These differences are not merely procedural; they reflect distinct philosophies, organizational structures, and priorities that profoundly impact development strategies, timelines, and costs.
Divergent Regulatory Philosophies and Structures
The fundamental organizational structures and underlying philosophies of the FDA and the EU regulatory bodies represent a significant divergence, shaping how drugs and devices are evaluated and approved.
Centralization vs. Multi-Stakeholder Coordination
The FDA operates as a single, centralized governmental authority, overseeing all drug and device approvals within the United States . This centralized structure offers inherent advantages, such as common rules applied uniformly across the nation and a potentially unified, streamlined decision-making process. For manufacturers, this can translate into a more predictable regulatory environment, as they interact primarily with one agency.
In stark contrast, the European Union’s regulatory system is characterized by a multi-stakeholder, coordinated approach. For drugs, the EMA coordinates centralized procedures that are valid across all member states, but national competent authorities still play a role, particularly for national or decentralized procedures . For medical devices, the EU system relies heavily on a decentralized network of independent Notified Bodies, which are designated by individual EU Member States . This multi-stakeholder approach means that while there are overarching EU regulations (MDR, IVDR), national laws and regulations may still apply at the country level. Furthermore, different Notified Bodies, despite being recognized across the EU, may have slightly different interpretations of the regulations or varying levels of expertise and capacity .
The EU’s decentralized, multi-stakeholder approach, while designed to foster harmonization across diverse member states and accommodate national specificities, inherently introduces greater complexity and potential for variability compared to the FDA’s centralized authority. This distributed responsibility, while a strength in accommodating diverse national contexts, can lead to “varying approval timelines and requirements, as different Notified Bodies may have slightly different interpretations of the regulation”. This variability can complicate strategic planning for companies, making the EU market entry process less straightforward and potentially less predictable than navigating the FDA’s single, unified system.
Risk-Benefit Paradigms: A Tale of Two Approaches
The underlying philosophy guiding regulatory decisions in the US and EU, particularly concerning risk-benefit assessments, reveals distinct approaches that influence data requirements and approval outcomes.
The FDA historically developed as a consumer protection agency, firmly rooted in a “safe and effective” standard for both drugs and devices . This principle often translates into a demand for robust clinical evidence to demonstrate both safety and efficacy, particularly for high-risk devices . For drugs, the FDA tends to show a greater tolerance for uncertainty in benefit-risk assessments, often relying more frequently on surrogate endpoints and limited clinical data, especially within its accelerated approval pathways . This pragmatic approach aims to expedite access for patients with serious unmet medical needs, with a commitment to gather more definitive data post-market.
Conversely, the EU’s regulations for drugs, while also aiming for a positive risk-benefit balance for the target population , have historically shown a stronger focus on long-term safety and public health priorities . For medical devices, the EU’s regulatory framework, stemming from a need to harmonize inter-state commercial interests while preserving national autonomy, initially focused primarily on a “safe” standard . However, the new Medical Device Regulation (MDR) has significantly elevated the bar, explicitly requiring manufacturers to provide comprehensive clinical evidence to demonstrate not just safety, but also performance and effectiveness. This means the MDR demands a higher quantity and quality of clinical data compared to previous directives .
The evolving EU regulatory philosophy, particularly under the MDR, is converging towards a “safe and effective” standard for devices, similar to the FDA. This signifies a global trend towards higher evidence thresholds for market authorization. While the FDA’s willingness to use surrogate endpoints for drugs allows for faster initial approvals, the EMA’s approach, sometimes based on more mature data, can lead to broader indications or a more robust long-term safety profile . This philosophical divergence means companies must carefully tailor their evidence generation strategies to meet the specific expectations of each regulatory body, rather than assuming a one-size-fits-all approach.
Approval Timelines and Success Rates: A Race to Market?
The speed and likelihood of regulatory approval are critical factors for pharmaceutical and medical device companies, directly impacting market entry, revenue generation, and competitive positioning.
Drugs: FDA’s Expedited Edge vs. EMA’s Comprehensive Review
When comparing drug approval timelines, the FDA generally demonstrates a faster review process than the EMA. Studies indicate that for novel drugs approved by both agencies, the median FDA review time was shorter, with a median difference of 121.5 days. This lag in the EU includes the approximately 60 days required for the European Commission to grant the marketing authorization after the EMA’s Committee for Medicinal Products for Human Use (CHMP) issues a positive opinion. A significant contributor to this difference is the FDA’s more frequent utilization of expedited programs like Fast Track, Priority Review, and Accelerated Approval . For drugs that underwent FDA expedited programs, review times were even shorter when compared to the same drugs approved by the EMA through its standard procedure, showing a median difference of 138 days .
Between 2013 and 2023, the FDA authorized significantly more novel drugs (583) than the EMA (424) . The FDA also granted a considerably higher number of exclusive drug approvals (185) compared to the EMA (42) during this period . This suggests that the US market often sees new drugs introduced earlier and more frequently.
However, a joint EMA/FDA analysis of 107 new drug applications submitted between 2014 and 2016 revealed a high degree of alignment, with the two agencies agreeing on approvals more than 90% of the time . When divergences did occur, differences in conclusions about efficacy were the most common reason . Notably, in instances where the EMA approved drugs that the FDA initially declined, it was often because the EMA reviewed applications that included “additional clinical trials or, particularly for oncology medicines, more mature data from the same clinical trial” . This more comprehensive data often led the EMA to grant standard approval, a broader indication, or approval for use as first-line therapy. For example, the COVID-19 vaccine Spikevax was authorized by the EMA in December 2021, while the FDA authorized it in January 2022 . In 2024, the FDA approved 50 novel drugs compared to the EMA’s 46 . While both approved Pfizer’s Hympavzi for hemophilia and Ipsen’s Iqirvo and Gilead’s Livdelzi for primary biliary cholangitis, significant differences emerged in neurological indications. The FDA approved Kisunla (donanemab) for Alzheimer’s in July 2024, which was still under EMA review. Conversely, the EMA approved Eisai and Biogen’s Leqembi (lecanemab) in late 2024, over a year after FDA approval .
This scenario presents a strategic trade-off for companies: faster initial market entry in the US, particularly through expedited programs, can allow for quicker recouping of R&D costs . However, the EMA’s slower, more comprehensive review, sometimes based on more mature data, can lead to broader indications or a more robust long-term safety profile upon EU approval . This influences global market strategies and patient access, as some effective treatments may be available in the US for months before reaching Europe.
Devices: US Speed vs. EU’s Enhanced Scrutiny
Bringing a medical device to market typically takes an average of 3 to 7 years. Historically, the European Union often saw the first approval of medical devices. Between 2005 and 2010, 63% of devices approved in both the US and EU were approved first in the EU. This trend was partly attributable to the EU’s less rigorous “safe” standard for devices, in contrast to the FDA’s “safe and effective” requirement .
However, this earlier market access in the EU came with a trade-off. Devices approved first in the EU were associated with a significantly increased risk of post-marketing safety alerts and recalls (27% for EU-first approvals compared to 14% for US-first approvals) . This raised concerns about the sufficiency of pre-market evidence in the EU system .
The implementation of the new EU Medical Device Regulation (MDR) has fundamentally reshaped this dynamic. The MDR has significantly increased regulatory requirements, demanding more extensive clinical evidence and robust post-market surveillance for devices . This heightened scrutiny, particularly concerning the Clinical Evaluation Report (CER) and Post-Market Clinical Follow-up (PMCF), has led to manufacturers experiencing longer approval timelines under the EU-MDR.
Consequently, a notable shift in strategic market entry has occurred. Many manufacturers now opt to pursue FDA approval first for medical devices to gain market entry more quickly, before tackling the more demanding EU-MDR process . This preference for initial US approval is often driven by the potentially faster and more cost-effective 510(k) and De Novo pathways for moderate-risk devices in the US. The shift in EU medical device regulation from a “safe” to a more “safe and effective” standard, coupled with increased Notified Body scrutiny, has effectively reversed the historical trend of faster EU market entry, now pushing manufacturers towards initial US approval for speed. This profoundly impacts global commercialization strategies, as companies prioritize markets that offer quicker time-to-revenue and the ability to generate early real-world data to support subsequent, more rigorous EU submissions.
Data Requirements and Clinical Evidence: The Foundation of Approval
The bedrock of any regulatory approval is the comprehensive data package demonstrating a product’s safety, efficacy, and quality. While both the FDA and EMA share this fundamental requirement, nuances in their expectations for preclinical, clinical, and post-market data can significantly influence development strategies.
Preclinical and Clinical Data Expectations
Both the FDA and EMA mandate extensive preclinical data before human trials can commence. This includes detailed toxicology studies to identify adverse effects, pharmacokinetics (ADME) studies to understand drug movement in the body, and pharmacology studies to determine biological activity . These studies must be conducted under strict Good Laboratory Practice (GLP) guidelines to ensure data credibility and reproducibility .
In the clinical trial phases (Phase I, II, and III), both agencies require a systematic assessment of safety and efficacy in human subjects . However, subtle yet significant differences can emerge in the quantity and maturity of clinical data expected at the time of submission. The FDA, particularly through its expedited approval pathways, may show a greater willingness to rely on surrogate endpoints (e.g., biomarkers) and limited clinical data to grant earlier market access . This pragmatic approach allows for quicker availability of therapies for serious conditions, with the understanding that confirmatory data will be gathered post-market .
In contrast, the EMA, while also having expedited pathways, has historically shown a preference for more comprehensive evidence. When reviewing applications, the EMA may often consider “additional clinical trials or, particularly for oncology medicines, more mature data from the same clinical trial” compared to what was submitted to the FDA . This can lead to divergent conclusions regarding efficacy, as the EMA may require a higher level of certainty before granting approval or a broader indication .
For medical devices, the EU Medical Device Regulation (MDR) has dramatically increased the quality and quantity of clinical data required compared to its predecessor, the Medical Device Directive (MDD) . The Clinical Evaluation Report (CER) has become a cornerstone, demanding robust clinical evidence for all device classes if clinical claims are made, a departure from the previous, less stringent requirements. This contrasts with some Class I and II devices in the US that may be exempt from extensive clinical data requirements, relying instead on general controls or substantial equivalence to a predicate. The MDR’s heightened emphasis on the CER and robust clinical evidence, even for lower-risk devices, signifies a fundamental shift towards a more “drug-like” evidence standard for devices in Europe, impacting development costs and timelines.
The Growing Importance of Post-Market Data Collection
A clear trend across both regulatory landscapes is the increasing emphasis on post-market data collection. This reflects a growing recognition that pre-market clinical trials, while essential, cannot fully capture all real-world safety and efficacy nuances, especially for rare adverse events or long-term outcomes. This evolution points towards a lifecycle approach to product safety and effectiveness.
The FDA mandates Phase IV studies for drugs to monitor long-term safety, rare side effects, and use in special populations . For medical devices, particularly those approved via accelerated pathways or those with remaining uncertainties, the FDA can require specific post-market studies to gather additional real-world evidence. The Sentinel Initiative is a prime example of the FDA’s commitment to leveraging real-world data for continuous safety monitoring.
Similarly, the EMA requires a Risk Management Plan (RMP) for all new drugs, outlining how risks will be managed throughout the product’s lifecycle. For medical devices, the EU MDR has significantly strengthened post-market surveillance (PMS) and introduced mandatory Post-Market Clinical Follow-up (PMCF) for all device classes, including legacy devices . PMCF involves continuous, proactive data collection from the use of CE-marked devices in humans to confirm safety and performance, identify unknown side effects, and ensure the ongoing acceptability of the benefit-risk ratio.
This increasing global emphasis on post-market data, particularly real-world evidence (RWE) and continuous monitoring, reflects a regulatory evolution towards a lifecycle approach to product safety and efficacy. This means that regulatory approval is no longer a static endpoint but the beginning of an ongoing commitment to data collection and risk management. For manufacturers, this necessitates investing in robust Quality Management Systems (QMS) and data infrastructure to support continuous monitoring, analysis, and reporting even after market entry.
The Economic Landscape: Costs of Development and Market Access
The financial realities of bringing drugs and devices to market are staggering, and understanding the cost structures in both the US and EU is crucial for strategic business planning.
Drug Development Costs: Billions at Stake
Developing a new prescription drug is one of the most expensive ventures in modern business. The average capitalized cost of developing a new prescription drug is approximately $2.6 billion, a figure that includes research, testing, regulatory approval, and the substantial costs of failed drugs that never make it to market. The entire process, from initial discovery to market approval, typically spans 10 to 15 years.
Breaking down these costs, preclinical research alone (before human trials) takes 3 to 6 years and costs between $300 million and $600 million. Clinical trials represent a major financial commitment:
Phase I: Costs between $1.5 million and $6 million per drug.
Phase II: Can range from $6 million to $20 million per drug.
Phase III: The most expensive phase, costing between $10 million and $40 million per drug.
These immense financial investments are coupled with a high risk of failure; only about 12% of drugs that enter clinical trials eventually receive FDA approval. The sheer scale of this investment underscores the critical need for strategic decisions around regulatory pathways to optimize return on investment and manage the high risk of failure. Faster approval pathways, like the FDA’s expedited programs, can reduce time-to-market, potentially allowing for earlier revenue generation and quicker recouping of these substantial R&D costs . Conversely, a longer, more stringent pathway, such as that sometimes seen with EMA approvals, might lead to a more robust label or broader indication, potentially justifying higher pricing or a larger market share in the long run.
Medical Device Development Costs: A Significant Investment
The financial investment required to develop and bring a medical device to market is also substantial, though generally lower than for drugs. The average cost to develop a medical device can reach upwards of $30 million for Class II 510(k) cleared devices, with approximately $24 million of this allocated to FDA-required activities, including clinical studies. For high-risk Class III devices, the costs can soar to $94 million. Interestingly, nonclinical development stages account for a significant portion (80.5% to 85%) of the overall capitalized development cost for therapeutic complex medical devices, while the FDA submission, review, and approval stage itself comprises a relatively small fraction, around 0.5%. This indicates that the bulk of the investment occurs in the R&D and preclinical/clinical testing phases, rather than the final regulatory review.
In the European Union, obtaining EU MDR certification is also a complex and costly procedure, with expenses varying significantly based on device complexity and classification. Implementing a robust Quality Management System (QMS) can range from €5,000 to €50,000, depending on the organization’s size and complexity. Notified Body assessments, which are compulsory for most device classes, can exceed €100,000, with costs varying based on the device class (e.g., Class IIb manufacturers may pay €10,000-€20,000, while Class III manufacturers could face costs as high as €50,000 for the assessment itself, plus annual fees). Clinical trials, if required, represent one of the most expensive aspects of acquiring MDR certification, with costs ranging from €50,000 to €500,000.
While the FDA approval stage for devices represents a small fraction of total development costs, the EU MDR’s increased stringency, particularly for clinical data and Notified Body involvement, is significantly elevating the cost of EU market access. This suggests that while initial development costs are high globally, the regulatory compliance costs for market entry are rising disproportionately in the EU due to the new regulations, influencing where companies might prioritize initial market launches.
Price Differentials and Reimbursement Challenges
Beyond the regulatory approval itself, the true market access and commercial success of a drug or device are heavily influenced by pricing and reimbursement policies, which exhibit significant disparities between the US and EU.
For prescription drugs, US prices, particularly for brand-name products, are substantially higher than in other developed countries. In 2022, US manufacturer gross prices for all drugs were nearly three times as high as prices in 33 OECD comparison countries. For brand-name originator drugs, US prices were 422% of prices in comparison countries, or at least 322% even after adjusting for estimated rebates . Conversely, US prices for unbranded generics were generally lower .
The price differentials for medical devices are even steeper. Prices for certain cardiac implant devices, for instance, have been found to be two to six times higher in the US compared to Germany. This stark contrast highlights fundamental differences in healthcare economics and reimbursement philosophies between the regions. The US operates more on a market-driven principle, allowing higher prices, while European countries often employ national-level Health Technology Assessment (HTA) bodies to evaluate the cost-effectiveness of new medical devices and drugs before they are reimbursed by public health systems.
This means that even after gaining regulatory approval from the EMA for drugs or achieving CE marking under the MDR for devices, companies must navigate a complex and fragmented web of national reimbursement processes in the EU. Each EU country has its own healthcare system and makes independent reimbursement decisions. Consequently, additional data, often beyond what was required for regulatory approval, might be necessary to satisfy HTA bodies and demonstrate economic value for reimbursement purposes. This adds another significant layer of complexity and cost to achieving full market access in Europe, requiring a dual strategic focus on both regulatory approval and reimbursement planning from the outset.
Leveraging Regulatory Intelligence for Competitive Advantage
In the increasingly complex and competitive global pharmaceutical and MedTech landscape, regulatory intelligence transcends mere compliance; it transforms into a strategic asset that can unlock significant competitive advantage.
Strategic Market Entry: Which Path First?
The decision of where to initiate market entry (the US or the EU) is a pivotal strategic choice, profoundly influenced by the distinct regulatory timelines, costs, and evidence requirements of each region.
For medical devices, a notable shift has occurred in recent years. Many manufacturers now strategically opt to pursue FDA approval first to gain market entry more quickly, before tackling the often more demanding EU-MDR process . This preference for the US as the initial market is frequently driven by the potentially faster and more cost-effective 510(k) and De Novo pathways available for moderate-risk devices in the US. The increased rigor and longer timelines associated with EU-MDR compliance have made the US a more attractive first-launch market for many device companies. This strategic sequencing of US and EU market entry for medical devices has profoundly impacted global commercialization strategies. It allows companies to achieve earlier time-to-revenue, establish competitive positioning, and generate valuable early real-world data that can then support subsequent, more rigorous EU submissions.
For drugs, while FDA approval is often faster, particularly through its expedited pathways, the EMA’s eventual approval might come with broader indications or a more robust label, often because the EMA has reviewed more mature or additional clinical data . Companies must carefully weigh the benefits of early market access in the US—which can facilitate quicker recouping of substantial R&D investments—against the potential for a more comprehensive and perhaps more enduring market position in the EU with a broader label, even if it means a slightly longer wait. This strategic trade-off requires a nuanced understanding of market dynamics, patient needs, and competitive landscapes in both regions.
Optimizing R&D and Clinical Development Strategies
Regulatory intelligence is instrumental in optimizing R&D and clinical development strategies, ensuring that efforts are not only compliant but also efficient and globally aligned. By deeply understanding the requirements of multiple jurisdictions, companies can design preclinical studies and clinical trial protocols that, where possible, meet the standards of both the US and EU simultaneously . This “global by design” approach can significantly reduce the need for redundant studies, thereby saving considerable time and resources.
Engaging with regulatory agencies early in the development process is a best practice that can yield substantial benefits. For the FDA, this involves utilizing the Q-Submission (Pre-Sub) process, which provides an opportunity to clarify regulatory requirements, discuss proposed study designs, and receive feedback on data gaps before formal submissions . Similarly, the EMA offers various opportunities for early dialogue and scientific advice, including “protocol assistance” for orphan medicines. This proactive engagement can clarify expectations, address uncertainties, and streamline the entire regulatory process, ultimately cutting down the time and cost related to pivotal studies.
For drugs, a clear understanding of the FDA’s expedited pathways means designing clinical trials to generate data suitable for these accelerated routes, which can significantly shorten overall development times and accelerate patient access. For medical devices, aligning device classification with clinical claims, labeling, and future reimbursement strategies from the outset is crucial . This integrated approach ensures that the evidence generated throughout development serves not only regulatory approval but also market access and commercial success.
Harnessing Patent Data for Business Intelligence: The Role of DrugPatentWatch
In the intensely competitive pharmaceutical landscape, intellectual property (IP) is a paramount asset, and patent data offers a rich vein of business intelligence. Tools like DrugPatentWatch provide deep knowledge on pharmaceutical drugs, encompassing patents, suppliers, generics, formulation details, and critical litigation information . This platform offers a fully integrated database that allows for freeform searching and dynamic browsing of all types of data pertaining to pharmaceuticals and their associated patents, covering both US and international jurisdictions.
DrugPatentWatch provides comprehensive data on patent expirations, clinical trials, Paragraph IV challenges (related to generic drug applications), and top patent holders. This wealth of information is invaluable for various stakeholders across the pharmaceutical value chain:
For Branded Pharmaceutical Manufacturers: The platform enables the assessment of past successes of patent challengers and helps elucidate the research paths of competitors. By monitoring patent landscapes, branded companies can anticipate generic entry, plan life-cycle management strategies, and identify potential competitive threats, thus protecting their market share and maximizing exclusivity.
For Generic Drug Manufacturers and API (Active Pharmaceutical Ingredient) Suppliers:DrugPatentWatch is a powerful tool for identifying market entry opportunities. It allows for the prediction of branded drug patent expiration dates and the identification of potential generic suppliers, which is crucial for portfolio management and strategic investment decisions.
For Buyers, Wholesalers, and Distributors: This intelligence helps in preventing overstock of branded drugs as they approach patent expiration, enabling more efficient inventory management and procurement strategies.
By integrating patent intelligence from platforms like DrugPatentWatch with a robust regulatory strategy, companies gain a holistic view of the market. This combined approach enables them to anticipate market shifts, identify competitive threats, and proactively manage their product portfolios, moving beyond mere regulatory compliance to a truly proactive and strategic market positioning. The platform’s ability to provide global biopharmaceutical business intelligence, including data on drugs in development and litigation, empowers users to make better and more informed business decisions, identify future therapeutic indications, and set up daily email alert watch lists for critical market changes.
The Horizon: Emerging Trends and Future Regulatory Evolution
The regulatory landscape for drugs and medical devices is not static; it is a dynamic environment constantly evolving in response to rapid technological advancements, shifting public health priorities, and the increasing globalization of pharmaceutical and medical technology industries.
The Transformative Impact of Artificial Intelligence (AI) and Machine Learning (ML)
Artificial Intelligence (AI) and Machine Learning (ML) are rapidly reshaping nearly every facet of drug development, clinical trials, diagnostics, and pharmaceutical manufacturing. These technologies hold immense promise for accelerating drug discovery, optimizing processes, and enhancing safety monitoring, with estimates suggesting AI could generate $60 to $110 billion annually in economic value for the pharma and medical-product industries .
Applications of AI/ML in Pharma and MedTech:
Drug Discovery and Preclinical Testing: AI-driven tools can rapidly analyze vast chemical, genomic, and proteomic datasets to identify promising drug candidates, predict molecular behavior, and assess drug-likeness and target-binding affinities with greater speed and accuracy than traditional methods . AI also plays a growing role in simulating in vivo biological systems, enabling researchers to predict pharmacokinetics and toxicity without immediate reliance on extensive animal models, potentially shortening preclinical development timelines . For example, an AI-designed drug candidate by Insilico Medicine reached human clinical trials within 18 months, significantly faster than standard preclinical periods.
Clinical Trial Design and Management: AI algorithms are increasingly utilized to optimize patient stratification, recruitment, and adherence monitoring in clinical trials . Natural Language Processing (NLP) tools can analyze trial protocols and outcomes to identify best practices and refine future trial designs .
Pharmacovigilance and Post-Market Surveillance: AI enhances the monitoring of drug safety by automatically detecting adverse drug events (ADEs) from electronic health records, social media, and patient forums . In manufacturing, AI/ML can optimize batch production, enable predictive maintenance, improve process control, and facilitate real-time quality monitoring .
Regulatory Responses and Challenges:
Both the FDA and EMA are actively addressing the regulatory implications of AI and ML. The FDA has issued draft guidance on using AI to support regulatory decision-making for drugs and biological products and is prioritizing the deployment of a unified, secure AI system across its centers . The EMA published a Reflection Paper on AI in the medicinal product lifecycle, emphasizing a risk-based approach and encouraging comprehensive assessment for high-impact AI systems . AI systems used in quality control or process control within pharmaceutical manufacturing are generally classified as “high-risk” under the EU AI Act, requiring robust risk assessments, human oversight, and transparency measures.
However, the integration of AI/ML, while promising unprecedented efficiency, introduces complex regulatory challenges that necessitate a paradigm shift in regulatory oversight and industry compliance:
Validation and Verification: Traditional validation approaches are often inadequate for adaptive AI/ML algorithms, whose behaviors may change over time . Regulators advocate for “locked” models with predefined change control plans for updates, and continuous learning models are viewed skeptically unless robust tracking mechanisms exist . This requires the development of “dynamic validation” methodologies .
Data Integrity: Ensuring data traceability and upholding ALCOA+ principles (Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, Available) throughout the AI data pipeline is critical . Black-box algorithms can obscure data provenance, necessitating explainable AI (XAI) techniques .
Explainability and Transparency: The inherent difficulty in deciphering the internal workings and conclusive derivations of complex AI models poses a significant challenge . Regulators expect manufacturers to understand the logic behind AI predictions and provide scientific justifications, pushing for “Explainability by Design” .
Bias and Representativeness: The potential for bias and unreliability introduced by variations in training data quality, volume, and representativeness is a key concern, as biased AI models could lead to inequitable outcomes or safety issues .
Model Drift: The susceptibility of AI model performance to change over time or across different operational environments underscores the necessity for continuous lifecycle maintenance and monitoring .
The integration of AI/ML into drug and device development, while holding immense promise for efficiency, fundamentally challenges traditional regulatory principles of control, reproducibility, and traceability. This forces regulators to develop new frameworks and for industry to invest in new compliance strategies, effectively creating a new frontier of regulatory science that will shape future approvals.
Digital Health Technologies (DHTs): A New Frontier in Regulation
Digital Health Technologies (DHTs), encompassing a broad range of hardware and/or software such as mobile phones, smartwatches, sensors, and mobile applications, are rapidly transforming healthcare and revolutionizing clinical trials . These technologies enable remote data acquisition, offering richer and more continuous datasets from participants in their home environments, potentially reducing the need for frequent site visits and increasing diversity in trials .
DHTs can function as standalone medical devices, often categorized as Software as a Medical Device (SaMD), or they can be used as tools to collect data for medicinal product development . Examples include imaging systems using algorithms for skin cancer diagnosis, smart sensor devices estimating heart attack probability , or wearable devices collecting health-related parameters for clinical studies .
Both the FDA and EMA are actively engaged in regulating this new frontier:
FDA’s Approach: The FDA has established a Digital Health Center of Excellence (DHCoE) within its Center for Devices and Radiological Health (CDRH) to foster responsible innovation and provide scientific expertise on DHTs . The DHCoE issues guidance documents on various aspects, including SaMD, AI/ML-enabled devices, cybersecurity in medical devices, and recommendations for remote data acquisition in clinical investigations. The FDA also provides a “Digital Health Policy Navigator” to help developers understand how policies apply to their products and determine regulatory oversight. The agency has qualified DHTs under its Medical Device Development Tools program, such as a biomarker to evaluate atrial fibrillation burden .
EMA’s Approach: The EMA has also recognized the regulatory utility of DHT-derived endpoints in drug development. A landmark example is the qualification of the stride velocity 95th centile as a primary endpoint for ambulatory Duchenne Muscular Dystrophy studies, demonstrating the agency’s acceptance of such novel data sources . The EMA’s qualification process assesses whether data derived by a DHT are acceptable to support a Marketing Authorisation Application .
The rapid proliferation of Digital Health Technologies is blurring traditional regulatory boundaries, demanding integrated regulatory approaches and increased collaboration between device and drug regulatory bodies. Because DHTs can serve dual purposes—as medical devices themselves or as data collection tools for drugs—they often fall under both medical device and medicinal product regulatory frameworks . This overlap necessitates “increased collaboration between various stakeholders, including regulators (medicines regulators and device bodies)” and the development of “integrated evaluation pathway[s]” for complex products . This trend indicates that future regulatory success for DHTs will increasingly depend on navigating a complex, interconnected web of rules and agencies, requiring manufacturers to understand and comply with both sets of regulations.
Global Harmonization Efforts and Persistent Challenges
The pharmaceutical and medical device industries are inherently global, making regulatory harmonization a critical goal. International regulatory harmonization aims to align technical requirements for product development and marketing, with the overarching objectives of reducing unnecessary duplication of clinical testing, promoting efficiency, and ensuring early patient access to medicinal products.
Several international organizations actively facilitate these efforts:
International Council for Harmonisation (ICH): ICH brings together regulatory authorities and the pharmaceutical industry to harmonize scientific and technical aspects of drug registration. Its mission is to achieve greater harmonization to ensure that safe, effective, and high-quality medicines are developed and registered in the most resource-efficient manner.
International Pharmaceutical Regulators Programme (IPRP): IPRP provides a multilateral forum for regulatory authorities to exchange information, advance harmonization projects, and promote regulatory best practices.
Pharmaceutical Inspection Co-operation Scheme (PIC/S): PIC/S is an informal co-operative arrangement focused on harmonizing Good Manufacturing Practice (GMP) inspection procedures worldwide, developing common standards, and providing training for inspectors.
Despite these concerted efforts and the clear benefits of harmonization, significant differences persist between the US and EU regulatory landscapes. These divergences stem from fundamental differences in regulatory philosophy, economic incentives, and national public health priorities:
Regulatory Philosophy: As discussed, the FDA and EMA have distinct approaches to risk management (FDA’s targeted REMS versus EMA’s mandatory RMP for all new drugs) . The FDA historically developed as a consumer protection agency, while EU regulations arose from a need to harmonize inter-state commercial interests while preserving national “autonomy”.
Financial Frameworks and Economic Incentives: Differences in financial frameworks and strategic marketing decisions of drug companies contribute to divergent approvals . The US often allows higher drug and device prices, creating different market dynamics and incentives compared to the EU, where national HTA bodies play a significant role in reimbursement decisions .
Therapeutic Priorities: While both agencies prioritize areas like oncology and infectious diseases, there can be subtle differences in therapeutic focus .
Geographical Factors: The inherent complexity of coordinating regulations across 28 diverse EU Member States, compared to the FDA’s single national authority, can lead to variations in interpretation and implementation .
Specific Case Examples: The COVID-19 pandemic highlighted these persistent differences, with the Spikevax vaccine authorized by the EMA in December 2021 and by the FDA in January 2022 . For medical devices, specific products like joint prostheses are classified differently in the US and the EU . The EU’s MDR has also significantly increased regulatory requirements, leading to different market access strategies for devices . Furthermore, a study found that more than one-fifth of drugs approved by the FDA between 2017 and 2020 were either refused marketing authorization or not recommended for public reimbursement in other countries (Australia, Canada, UK) due to unfavorable benefit-risk profiles or uncertain clinical benefits, often drugs initially approved by the FDA under expedited pathways .
While global harmonization efforts are ongoing and valuable, fundamental differences in regulatory philosophy, economic incentives, and national public health priorities continue to create persistent divergences. This necessitates tailored global market access strategies rather than a “one-size-fits-all” approach. Companies must continuously adapt to regional nuances, engage in early dialogue with regulatory bodies, and strategically plan their evidence generation to meet the specific demands of each market.
Conclusion: Charting a Course for Global Success
Navigating the complex regulatory landscapes of the United States and the European Union for drugs and medical devices is a formidable challenge, yet it is also a profound opportunity for strategic advantage. This analysis has illuminated that while both the FDA and the EMA/EU regulatory bodies share the overarching goal of ensuring patient safety and product efficacy, their distinct organizational structures, regulatory philosophies, and procedural nuances create significant differences in approval processes, timelines, costs, and data expectations.
The FDA’s centralized authority often allows for faster drug approvals, particularly through its expedited pathways, and has become the preferred initial market for many medical devices due to the increased stringency of the EU MDR. This speed, however, is balanced by a robust post-market surveillance framework, implicitly shifting some evidence-gathering to the post-approval phase. In contrast, the EU’s multi-stakeholder system, while offering flexibility through various approval pathways, often entails longer review times for drugs, sometimes based on more mature data leading to broader indications. For devices, the EU MDR’s heightened demands for clinical evidence and the pivotal role of Notified Bodies have significantly increased the regulatory burden and associated costs.
The integration of cutting-edge technologies like Artificial Intelligence and Digital Health Technologies is further reshaping these landscapes, introducing new regulatory challenges around validation, data integrity, and explainability. Despite ongoing global harmonization efforts, fundamental differences in regulatory philosophy, economic incentives, and national public health priorities continue to drive persistent divergences.
For business professionals in the pharmaceutical and medical device sectors, success in this global arena hinges on a deep, nuanced understanding of these dynamics. It requires a strategic, adaptive planning approach that considers:
Tailored Market Entry: Deciding which market to target first based on product type, risk profile, and the latest regulatory shifts.
Optimized R&D and Clinical Development: Designing global clinical trials that can satisfy multiple regulatory requirements, and engaging in early, proactive dialogue with agencies.
Comprehensive Regulatory Intelligence: Leveraging tools like DrugPatentWatch to integrate patent data with regulatory insights, anticipating market shifts, and identifying competitive opportunities.
Anticipating Future Trends: Staying abreast of evolving regulations for AI, Digital Health, and other emerging technologies to proactively adapt development and compliance strategies.
Ultimately, mastering the intricacies of US and EU approval processes is not merely about ticking boxes; it is about strategically charting a course that minimizes risk, optimizes investment, accelerates patient access, and secures a lasting competitive edge in the global healthcare market.
Key Takeaways
Drug and device development is a multi-billion dollar, multi-year endeavor with low success rates, making regulatory strategy critical for competitive advantage.
The FDA operates as a centralized authority, offering potentially faster drug approvals via expedited pathways and becoming a preferred initial market for many medical devices due to the EU MDR’s increased rigor.
The EU’s multi-stakeholder system (EMA, Notified Bodies) offers flexible drug approval pathways but can entail longer review times, sometimes leading to broader indications based on more mature data.
The EU Medical Device Regulation (MDR) has significantly increased clinical evidence requirements and the role of Notified Bodies, elevating EU device market entry costs and complexity.
Both the US and EU emphasize robust post-market surveillance, reflecting a lifecycle approach to product safety and efficacy, requiring continuous data collection and risk management from manufacturers.
Significant price disparities exist for drugs and devices between the US and EU, driven by differing healthcare economic models and reimbursement philosophies, necessitating distinct market access strategies.
Artificial Intelligence and Digital Health Technologies are transforming development, but introduce complex regulatory challenges around validation, data integrity, and explainability, requiring new compliance paradigms.
Despite global harmonization efforts, fundamental differences in regulatory philosophy and national priorities persist, demanding tailored, rather than uniform, global market access strategies.
Leveraging comprehensive regulatory intelligence, including patent data from platforms like DrugPatentWatch, is essential for anticipating market shifts, optimizing R&D, and securing competitive positioning.
Frequently Asked Questions (FAQ)
Why does the FDA often approve drugs faster than the EMA, and what are the implications for market entry strategy? The FDA often approves drugs faster due to its centralized structure and more frequent use of expedited programs such as Fast Track, Priority Review, and Accelerated Approval . These programs allow for quicker review times, sometimes by relying on surrogate endpoints or rolling submissions . The implication for market entry strategy is that companies may prioritize initial US approval to achieve faster time-to-revenue and recoup R&D costs more quickly . However, the EMA’s typically longer review, sometimes with more mature data, can lead to broader indications or a more robust long-term safety profile upon EU approval, influencing subsequent market expansion strategies .
How has the EU MDR changed the game for medical device manufacturers, particularly concerning clinical evidence? The EU Medical Device Regulation (MDR) has significantly raised the bar for medical device manufacturers. It transitioned from a largely “safe” standard to requiring comprehensive clinical evidence to demonstrate both safety and performance, akin to drug approval standards . This means manufacturers must now produce a detailed Clinical Evaluation Report (CER) with robust clinical data for all device classes, even Class I if clinical claims are made . The MDR also mandates continuous Post-Market Clinical Follow-up (PMCF) throughout a device’s lifetime . This increased stringency has led to longer approval timelines and higher compliance costs in the EU, causing many manufacturers to now pursue FDA approval first for quicker market access .
What are the primary financial implications of navigating both US and EU approval processes for a novel drug or device? Navigating both US and EU approval processes involves substantial financial implications. For drugs, average development costs are around $2.6 billion, with preclinical and clinical trials being major cost drivers. For devices, costs can range from $30 million for Class II to $94 million for Class III in the US. In the EU, MDR certification adds significant costs for Quality Management System implementation, Notified Body assessments (potentially exceeding €100,000 depending on device class), and additional clinical trials. Beyond approval, significant price differentials exist, with US drug and device prices often much higher than in the EU . Companies must also budget for complex, country-specific reimbursement processes in the EU, which may require additional data beyond regulatory approval to demonstrate economic value.
How are emerging technologies like AI and Digital Health impacting the regulatory landscape in the US and EU, and what should companies prepare for? AI and Digital Health Technologies (DHTs) are profoundly impacting regulation by accelerating drug discovery, optimizing clinical trials, and enhancing safety monitoring . Both the FDA and EMA are developing new guidance, emphasizing risk-based approaches and requiring robust validation, data integrity, and transparency for AI/ML systems . DHTs are blurring traditional regulatory boundaries, often falling under both medical device and medicinal product regulations, necessitating increased collaboration between these regulatory domains . Companies should prepare for evolving regulatory frameworks, complex validation requirements for adaptive algorithms, challenges in ensuring data integrity and explainability, and the need for tailored strategies that address the dual nature of many DHTs.
Despite harmonization efforts, what fundamental philosophical differences continue to drive divergent regulatory outcomes between the FDA and EMA/EU? Despite harmonization efforts by organizations like ICH, fundamental philosophical differences persist. The FDA operates as a centralized consumer protection agency, historically emphasizing “safe and effective” and often showing a greater tolerance for uncertainty in benefit-risk assessments, especially with expedited pathways . In contrast, the EU’s multi-stakeholder system, while aiming for harmonization across diverse member states, stemmed from a need to balance inter-state commercial interests with national autonomy. The EMA often shows a stronger focus on long-term safety and public health priorities for drugs , and the EU MDR has significantly elevated the evidence bar for devices. These differences in underlying philosophy, coupled with varying financial frameworks and national public health priorities, continue to lead to divergent approval timelines, data requirements, and market access strategies .
A decade comparison of regulatory decision patterns for oncology products to all other non‐oncology products among Swissmedic, European Medicines Agency, and US Food and Drug Administration, accessed July 23, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC10499418/
Comparison of regulatory approval system for medicines in emergency among Japan, the United States, the United Kingdom, Europe, and China, accessed July 23, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC11404807/
Regulatory landscape of accelerated approval pathways for medical devices in the United States and the European Union – PMC – PubMed Central, accessed July 23, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC12119601/
Food and Drug Administration vs European Medicines Agency: Review times and clinical evidence on novel drugs at the time of approval – PMC, accessed July 23, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC6983504/
Comparison of rates of safety issues and reporting of trial outcomes for medical devices approved in the European Union and United States: cohort study | The BMJ, accessed July 23, 2025, https://www.bmj.com/content/353/bmj.i3323
Regulatory considerations for successful implementation of digital endpoints in clinical trials for drug development – PMC – PubMed Central, accessed July 23, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC11882986/
Advancing Regulatory Oversight of Medical Device Trials to Align with Clinical Drug Standards in the European Union – PMC – PubMed Central, accessed July 23, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC12195702/
Navigating the Regulatory Pathway for Medical Devices—a Conversation with the FDA, Clinicians, Researchers, and Industry Experts – PubMed Central, accessed July 23, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC8920055/
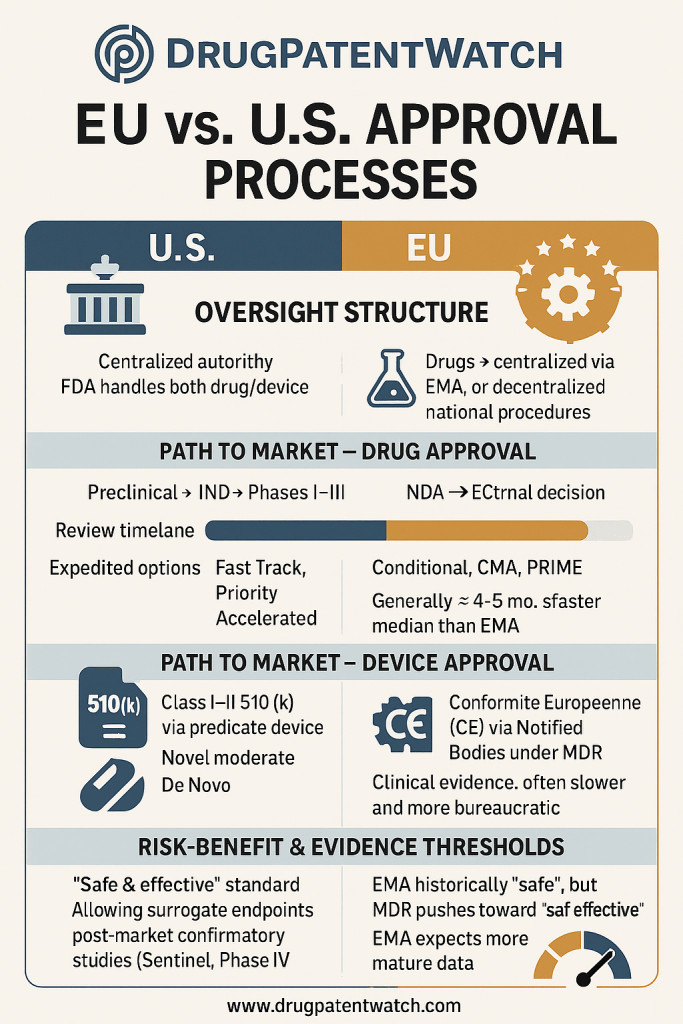


















(2), and generic competition")




(2) Regulatory Pathway for New Drug Applications: An Overview on the Advanced Formulation Approach and Challenges")


