The pharmaceutical and biotechnology industries operate within a complex ecosystem, where the successful launch of a generic drug hinges on a confluence of scientific rigor, regulatory compliance, and strategic foresight. This report provides an in-depth analysis of the critical factors that collectively predict the likelihood of successful Abbreviated New Drug Application (ANDA) approval, transforming the understanding of regulatory, scientific, and strategic nuances. The objective is to empower generic drug manufacturers and related industry professionals to navigate this intricate landscape with enhanced precision and confidence, ultimately contributing to broader public health and economic benefits.
The Generic Drug Landscape: A Strategic Overview
Generic drugs are more than just cost-effective alternatives; they are fundamental pillars of healthcare affordability and accessibility worldwide. Their ability to provide clinically equivalent options, often at prices 30% to 80% lower than their brand-name counterparts, significantly alleviates the financial strain on patients and healthcare systems.1 This economic advantage has propelled the generic pharmaceuticals market into a period of substantial growth, with projections indicating a valuation of USD 682.9 billion by 2030.1 This trajectory is largely fueled by the continuous expiration of patents on numerous high-value branded medications, underscoring a global reliance on generics to manage escalating healthcare expenditures.1
The economic impact of generic drugs extends far beyond immediate cost savings, contributing profoundly to the sustainability of broader healthcare systems. For instance, generic drugs saved the U.S. healthcare system an estimated $2.2 trillion from 2009 to 2019.2 This substantial financial relief enables healthcare systems to reallocate resources towards other critical areas, such as research and development for novel therapies, infrastructure enhancements, or the expansion of public health initiatives. This systemic financial benefit creates a compelling impetus for governments and regulatory bodies to actively promote and streamline the market entry of generic drugs.1 The growth trajectory of the generic market is thus inextricably linked to the efficiency and predictability of the ANDA approval process. Any bottlenecks or inefficiencies within this pathway directly impede market expansion, limit competition, and invariably keep drug prices elevated. Therefore, optimizing the ANDA process, through initiatives like the Generic Drug User Fee Amendments (GDUFA), is not merely a regulatory compliance exercise but a core strategic imperative that directly influences market dynamics and competitive positioning for generic manufacturers.1
Demystifying the Abbreviated New Drug Application (ANDA) Process
The Abbreviated New Drug Application (ANDA) pathway, a cornerstone established by the Hatch-Waxman Act of 1984, allows generic manufacturers to obtain FDA approval without replicating the extensive and costly clinical trials typically required for new drugs.3 Instead, the central requirement for an ANDA is to demonstrate that the proposed generic product is therapeutically equivalent to an already approved Reference Listed Drug (RLD).4 This demonstration of therapeutic equivalence necessitates that the generic drug matches the RLD across several critical parameters: active ingredients, conditions of use, method of administration, dosage form, strength, potency, and labeling.4 Critically, it must also exhibit equivalent bioavailability and bioequivalence.4 While minor differences in inactive ingredients or container closure systems may be permitted, these variations must be rigorously proven not to affect the drug’s safety or efficacy.4
The “abbreviated” nature of the ANDA process, while offering a faster and more cost-effective route to market, simultaneously demands an exceptionally high degree of precision in demonstrating therapeutic equivalence.4 By eliminating the need for new clinical trials, the ANDA pathway significantly reduces development time and costs.4 However, this abbreviation shifts the regulatory burden entirely onto proving exact bioequivalence and pharmaceutical sameness. This means that even seemingly minor deviations in formulation, active ingredient purity, or pharmacokinetic behavior, if not meticulously justified and validated, can result in a Complete Response Letter (CRL) or outright rejection. The challenge for generic manufacturers transforms from demonstrating broad clinical efficacy to ensuring granular analytical and manufacturing fidelity, requiring unparalleled precision. Consequently, a comprehensive, multi-faceted analysis of the RLD, extending beyond its active ingredient, stands as an indispensable prerequisite for a successful ANDA submission.7 The ANDA process fundamentally relies on the safety and efficacy data of the RLD.8 This necessitates a deep dive into the RLD’s complete profile, including its chemical composition, precise formulation, detailed labeling, and regulatory history, as a critical “pre-ANDA preparation” step.4 This is not merely about copying the active ingredient; it involves understanding the subtle interplay of all components and their impact on the drug’s performance. Proactive and exhaustive RLD analysis ensures the proposed generic truly mirrors the original, minimizing unforeseen regulatory hurdles related to formulation or performance discrepancies.
The Competitive Arena: Seizing Opportunities in Generic Drug Development
The generic drug market is characterized by intense competition and dynamic shifts, demanding innovative strategies that extend beyond simple price leadership. Sustained success in this environment often requires the development of “higher-value generics”—products that offer improved efficacy, safety, or convenience through innovative formulations or delivery systems.9 Other key strategies include proactive anticipation of patent expirations, robust counter-strategies against brand-name company tactics, incentivizing generic uptake, employing effective marketing, exploring Over-the-Counter (OTC) formulations, launching authorized generics, and fostering strategic collaborations.9
Success in the generic market is increasingly shifting from a pure “copycat” model to one driven by strategic innovation and differentiation. While the core regulatory requirement for an ANDA is therapeutic equivalence, the market’s demand for “higher-value generics” 9 indicates that manufacturers must now invest in research and development (R&D) to offer enhanced product attributes, such as improved delivery systems or more patient-friendly formulations. This trend blurs the traditional lines between generic and innovative drug development, suggesting that sustained competitiveness requires a commitment to continuous improvement and targeted innovation, particularly for complex products. This necessitates a holistic, forward-looking business strategy that seamlessly integrates regulatory, legal, and commercial intelligence. The diverse strategies for success encompass regulatory filings (ANDA, bioequivalence), legal challenges (patent expiries, combating brand tactics), and commercial execution (marketing, OTC formulations).9 This complexity mandates a departure from siloed departmental approaches. Manufacturers must integrate these functions, leveraging tools like DrugPatentWatch for comprehensive patent intelligence 10 to anticipate market shifts, competitive maneuvers, and regulatory changes, ensuring that all efforts are synergistically aligned with overarching business objectives for market leadership.
Regulatory Foundations: The FDA’s Blueprint for ANDA Success
Meticulous Submission: The Cornerstone of Approval
A complete, accurate, and well-documented application serves as the absolute cornerstone for avoiding delays and ensuring a smooth review process.7 The FDA conducts a rigorous, multi-phase review that scrutinizes all submitted data, including bioequivalence and safety data, labeling compliance, and manufacturing site capabilities through inspections.4 The ANDA dossier must adhere to specific structural and content requirements, including a completed and signed application form, a detailed table of contents, and a clear, explicit reference to the RLD as the basis for submission.14
Active Ingredient and Formulation Identity: Beyond the Surface
The generic drug must be demonstrably identical to the RLD in its active ingredient(s), dosage form, strength, and route of administration.4 This fundamental requirement ensures that the generic delivers the same therapeutic effect. While minor differences in inactive ingredients are generally permissible, they come with a critical caveat: they must be rigorously proven not to affect the drug’s safety or efficacy.4 For specific dosage forms such as ophthalmic, otic, topical, or parenteral products, the FDA’s regulations are even more stringent, often requiring inactive ingredients to be “generally the same,” with limited, justified exceptions for components like preservatives or buffers.14
The seemingly minor details of inactive ingredients can escalate into significant regulatory hurdles, particularly for non-oral dosage forms, impacting approval likelihood. The FDA’s granular requirements for inactive ingredients, especially for specialized dosage forms 14, highlight a frequently overlooked area of potential deficiency. This implies that generic manufacturers cannot assume blanket flexibility regarding excipients. Instead, they must conduct exhaustive analysis and provide robust justification for any deviations, as these can subtly yet significantly impact the drug’s stability, performance, and safety profile.16 This is particularly critical for complex formulations where excipient-API interactions can influence bioavailability and degradation pathways.16 A profound understanding of the RLD’s complete formulation, including the precise role and characteristics of its inactive ingredients, is crucial from the earliest stages of generic development to prevent costly rework and delays. Given the FDA’s stringent scrutiny of inactive ingredients 14, the “reverse engineering” of the RLD’s formulation must extend beyond merely identifying the active pharmaceutical ingredient (API). Manufacturers must meticulously analyze the RLD’s excipients, their physical and chemical properties, and their contribution to the overall drug product performance and stability.16 This proactive, in-depth understanding enables informed decisions during generic formulation development, significantly minimizing the risk of late-stage deficiencies related to unexpected interactions or subtle non-equivalence caused by seemingly inert components.
Conditions of Use and Labeling: Precision in Communication
The proposed labeling for the generic drug must be substantively identical to the RLD’s currently approved labeling.14 Permissible differences are strictly limited to aspects such as manufacturer information, or the omission of patent/exclusivity-protected indications for which the generic is not seeking approval.14 A critical component of the submission is a side-by-side comparison of the proposed generic labeling with the RLD’s approved labeling, with all differences meticulously annotated and explained.14 This ensures transparency and facilitates FDA review.
Labeling, often perceived as a final-stage administrative task, is in fact a critical regulatory compliance checkpoint that can trigger immediate Refuse-to-Receive (RTR) decisions or subsequent Complete Response Letters (CRLs) if not managed with utmost precision.17 Errors or non-compliance in labeling are explicitly cited as common reasons for RTR decisions.17 This underscores that labeling is far from a mere formality; it is a precise regulatory requirement directly impacting the ANDA’s initial acceptance. Any deviation from the RLD’s approved labeling 14, unless specifically justified and clearly annotated, can halt the entire application process before substantive scientific review even begins. This highlights the necessity for regulatory affairs teams to be deeply involved in labeling development from the outset, ensuring strict adherence to FDA guidelines and the RLD’s profile. The “sameness” requirement for labeling extends to strategically excluding indications still under patent protection, necessitating continuous and sophisticated patent intelligence. The requirement that generic labeling match the RLD’s, “except for changes required because the drug product is not approved for a use for which the reference listed drug is approved” 14, directly links labeling compliance to intellectual property status. This implies that generic manufacturers must continuously leverage robust patent intelligence (e.g., from DrugPatentWatch) to accurately identify and carve out any indications still protected by method-of-use patents listed in the Orange Book.10 This demonstrates a sophisticated understanding of both regulatory requirements and the intricate intellectual property landscape.
Current Good Manufacturing Practices (cGMP): The Unwavering Standard
Compliance with Current Good Manufacturing Practices (cGMP) is a non-negotiable, fundamental requirement for obtaining pre-market approval for both new and generic drugs.20 The FDA rigorously assesses whether a manufacturing firm possesses the requisite facilities, equipment, and operational capabilities to consistently produce the drug it intends to market.20 The “Five Ps” of cGMP – Primary Materials and Products, Premises, People, Processes, and Procedures – comprehensively outline the core obligations for pharmaceutical manufacturers.22 These emphasize stringent quality assurance, proactive prevention of adulteration, and meticulous, ongoing documentation across all operational facets.22
cGMP compliance is not a static checklist but a dynamic, integrated quality system that underpins the entire product lifecycle, from development to post-market surveillance. The comprehensive nature of the “Five Ps” 22 illustrates that cGMP compliance permeates every aspect of a pharmaceutical operation, from the quality of raw materials to the training of personnel and the validation of manufacturing processes. This signifies that cGMP is not a one-time audit but an ongoing, continuous commitment to quality. Failures in cGMP can render a drug “adulterated” under the law 20, a severe regulatory violation. This highlights that robust, documented internal monitoring, auditing, and corrective actions are crucial not only for initial ANDA approval but also for maintaining post-market product quality and avoiding costly recalls.20 Proactive development and implementation of custom-tailored cGMP programs are essential to mitigate the significant risks of delays, rejections, and post-approval compliance issues.21 Given that manufacturing deficiencies are consistently identified as a leading cause of Complete Response Letters (CRLs) 23, generic manufacturers cannot afford a reactive approach to cGMP. Developing a bespoke, comprehensive cGMP program that includes thorough documentation, rigorous internal monitoring, and regular audits 21 is critical. This proactive stance ensures that manufacturing facilities and processes are “approval-ready” from the outset, significantly reducing the likelihood of FDA findings that can delay or outright prevent market entry, thereby protecting both investment and market opportunity.
Bioequivalence: The Scientific Mandate for Interchangeability
Bioequivalence studies form the scientific bedrock of generic drug approval, serving as the primary evidence that the generic drug will deliver the same therapeutic effect as the Reference Listed Drug (RLD).25 This rigorous demonstration scientifically justifies bypassing the need for new, extensive clinical trials for safety and efficacy.25
Pharmacokinetic Parameters: Unpacking Cmax, Tmax, and AUC
Bioequivalence studies are meticulously designed to compare the pharmacokinetic (PK) profiles of the generic drug with the innovator drug.25 The key PK parameters assessed are Cmax (maximum drug concentration in the bloodstream), Tmax (time to reach maximum concentration), and AUC (area under the concentration-time curve, representing total drug exposure over time).26 Cmax is crucial for assessing the rate and extent of drug absorption, Tmax indicates the speed of absorption, and AUC provides insights into the overall bioavailability.26 Comparable values across these parameters between the generic and RLD are essential for demonstrating therapeutic equivalence.26
Achieving bioequivalence is a precise scientific endeavor, where even slight deviations in pharmacokinetic profiles can be critical deal-breakers for approval. The intense focus on specific, quantifiable PK parameters (Cmax, Tmax, AUC) and the requirement for their precise measurement 26 underscore the exacting scientific rigor demanded by the FDA. It is not sufficient for a generic drug to merely feel or appear similar; it must behave identically within the human body. This implies that the design, execution, and statistical analysis of bioequivalence studies must be impeccable, as even marginal statistical deviations from the RLD’s PK profile can lead to a Complete Response Letter (CRL), irrespective of perceived clinical similarity.23 The inherent complexity of certain drug formulations, such as modified-release products or those with a narrow therapeutic index, significantly amplifies the challenge of demonstrating bioequivalence, often necessitating specialized and more intricate study designs.6 While bioequivalence is central to generic approval, its demonstration becomes considerably more challenging for complex formulations or drugs with narrow therapeutic windows.6 This suggests that a “one-size-fits-all” bioequivalence study design is inadequate. Generic manufacturers must tailor their study protocols to the unique characteristics of the RLD, potentially requiring more sophisticated, multi-dose, or prolonged studies to accurately capture the nuances of drug release, absorption, and systemic exposure, especially where small variations could have significant clinical consequences.
Rigorous Study Design and Statistical Validation: Meeting the 80-125% Confidence Interval
Bioequivalence studies are meticulously designed to evaluate the rate and extent of drug absorption, distribution, metabolism, and excretion.25 Typically, human volunteers receive both the test (generic) and reference (RLD) formulations at different times, with their blood levels rigorously monitored to assess drug behavior in the body.25 Rigorous statistical analysis is then employed to determine bioequivalence, with confidence intervals for the ratio of geometric means (often for Cmax and AUC) typically required to fall within the narrow range of 80% to 125%.30
The 80-125% acceptance criterion, while a numerical target, functions as a critical regulatory gatekeeper for generic market entry, demanding absolute precision. The 80-125% confidence interval 30 is more than a statistical benchmark; it is the FDA’s quantitative definition of acceptable therapeutic equivalence. Failing to meet this narrow window, even by a small margin, means the generic drug is not deemed therapeutically substitutable for the RLD. This emphasizes the paramount importance of meticulous study planning, robust statistical power, and flawless execution to ensure the generic’s pharmacokinetic profile falls squarely and consistently within this stringent acceptable range, directly impacting the likelihood of approval. Bioequivalence study challenges are a frequent and significant cause of ANDA delays and rejections, underscoring the critical need for meticulous planning and expert oversight from the outset.31 Bioequivalence study challenges are explicitly identified as intricate hurdles that require meticulous planning to minimize delays.31 This highlights that merely conducting a study is insufficient; its population selection, design, methodology, and statistical analyses must be “meticulously aligned with FDA expectations”.32 This implies that proactive engagement with experienced regulatory affairs experts and biostatisticians during the study design phase is a critical best practice to mitigate the substantial risk of costly delays or outright rejections stemming from methodological flaws or insufficient data.32
Dissolution Testing: A Critical Predictor of In Vivo Performance
Dissolution testing is a vital tool that measures the rate at which a drug is released from its dosage form (e.g., tablets, capsules, liquid suspensions).33 It is a mandatory requirement for all oral solid dose, oral semi-solid dose, and oral suspension products.33 This testing serves two crucial, interconnected functions: it is used during R&D to predict
in vivo bioavailability (the first step in understanding how the drug will be absorbed) and as an ongoing quality control (QC) tool during manufacturing.33 For drugs with low solubility (classified as BCS Class II and IV), bioavailability is primarily driven by their dissolution rate.33 Therefore, accurate and discriminatory dissolution methods are paramount for these products.33 Dissolution testing is particularly critical for Abbreviated New Drug Application (ANDA) filings, as generic products must demonstrate bioequivalence to the Reference Listed Drug (RLD).34 To simulate physiological conditions, dissolution is performed across the full pH range experienced by the dosage form as it transits through the gastrointestinal (GI) tract.34
Dissolution testing acts as an indispensable early warning system for potential bioequivalence issues, making it a critical and strategic R&D investment for generic manufacturers. The dual role of dissolution testing – predicting bioavailability and serving as a quality control measure 33 – elevates its status beyond a mere regulatory checkbox to a powerful developmental tool. By conducting dissolution tests across various pH conditions simulating the GI tract 34, manufacturers can gain predictive insights into how the drug will behave
in vivo. This early-stage intelligence is invaluable for identifying and rectifying potential formulation issues before committing to costly and time-consuming in vivo bioequivalence studies, especially for challenging formulations or low-solubility drugs, thereby directly influencing the likelihood of a successful ANDA. The FDA’s utilization of dissolution testing as a diagnostic tool for post-approval changes underscores its continuous importance throughout the drug’s lifecycle, extending beyond initial market approval.33 The FDA views dissolution testing as a “diagnostic tool to evaluate changes requested by manufacturers to approved products that may adversely impact bioavailability and product quality”.33 This perspective reveals that maintaining a consistent and approved dissolution profile is crucial not only for initial ANDA approval but also for ongoing compliance and avoiding post-market quality issues, such as recalls or adverse events. This implies that the robustness of dissolution methods developed during the ANDA process has long-term implications for the generic product’s sustained market presence and regulatory standing.
Analytical Method Validation: Ensuring Data Integrity and Reproducibility
Analytical Method Validation (AMV) is the meticulously documented process that demonstrates an analytical procedure’s suitability for its intended purpose.35 This rigorous process ensures that any method used consistently produces reliable, accurate, and reproducible results within specified parameters.35 Regulatory agencies worldwide, including the FDA, European Medicines Agency (EMA), and International Council for Harmonisation (ICH), mandate validated methods for the analysis of drug substances, drug products, and biological samples throughout all stages of drug development, manufacturing, and release . Key parameters rigorously assessed during AMV include accuracy (closeness to true value), precision (repeatability), specificity (ability to measure the analyte without interference), linearity (proportionality to concentration), range (reliable measurement interval), detection limit (LOD), quantitation limit (LOQ), and robustness (unaffected by small parameter variations) .
Analytical Method Validation is the fundamental bedrock upon which the integrity of all data submitted in an ANDA rests; deficiencies in AMV can invalidate entire sections of the application, regardless of the underlying science. If the analytical methods employed to generate critical data—such as that for bioequivalence studies, stability testing, impurity profiling, or content uniformity—are not rigorously validated, the reliability and trustworthiness of that data are fundamentally compromised . This means that even if a generic drug is, in fact, therapeutically equivalent, a flawed AMV can lead to a Complete Response Letter (CRL) because the FDA cannot rely on the submitted evidence.23 This implies that substantial investment in robust analytical capabilities and strict adherence to established validation protocols (e.g., ICH Q2(R1), FDA Guidance) are non-negotiable prerequisites for ANDA success, as they directly impact the credibility of the entire submission . The “robustness” parameter in Analytical Method Validation is critically important for predicting real-world manufacturing consistency and proactively mitigating post-approval quality issues. “Robustness,” defined as the method’s capacity to remain unaffected by small, anticipated variations in method parameters , is a crucial aspect of AMV. Manufacturing processes inherently involve minor variations. A robust analytical method ensures that quality control results remain accurate and reliable despite these small shifts, thereby guaranteeing consistent product quality. This directly impacts a manufacturer’s ability to reliably produce a high-quality product that consistently meets specifications, significantly reducing the risk of manufacturing-related CRLs or, worse, post-market recalls due to quality deviations.23
Table 1: Key Requirements for ANDA Submission (Regulatory & Scientific)
Section/Requirement Area
Specific Requirement/Guideline
Significance for Approval
Active Ingredients
Identical to RLD in chemical composition, strength, dosage form, route of administration 14
Ensures therapeutic equivalence and substitutability.4
Inactive Ingredients
Minor differences permissible, but must not affect safety/efficacy; stricter for non-oral forms 14
Prevents unexpected performance deviations or safety concerns.16
Conditions of Use
Proposed labeling conditions must be previously approved for RLD 14
Confirms the generic’s intended use aligns with established safety and efficacy.14
Labeling
Substantively identical to RLD’s approved labeling, with justified, annotated differences 14
Ensures clear, consistent communication to healthcare providers and patients; avoids RTRs/CRLs.17
Bioequivalence Studies
Demonstrate comparable PK profiles (Cmax, Tmax, AUC) to RLD, typically 80-125% CI 25
Scientific proof that the generic performs identically in the human body, justifying abbreviated pathway.25
Dissolution Testing
Mandatory for oral solids/suspensions; predicts bioavailability; QC tool; performed across GI pH range 34
Early indicator of in vivo performance; critical for low-solubility drugs and quality control.33
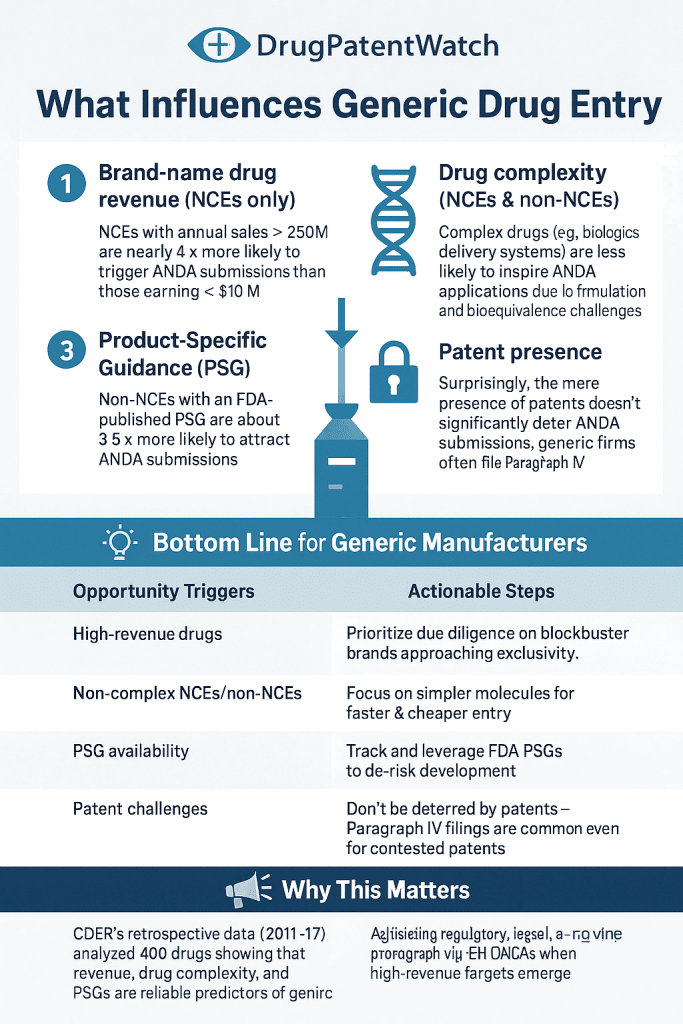


















(2) Pathway: Uncovering Drug Development Trends and Regulatory Requirements")


(2), and generic competition")




