This article was first published by Elinevan Overbeeke, Sissel Michelsen, Mondher Toumi, Hilde Stevens, Mark Trusheim, Isabelle Huys, and StevenSimoens in Drug Discovery Today under a Creative Commons License.
Highlights
• Challenges blocking market access of GTMPs are highly interrelated.
• Developers should seek support and early joint interactions with regulators and payers.
• Conditional marketing authorization and reimbursement mechanisms should be explored.
• RWE infrastructure and requirements should be developed on an international level.
• Efficient innovative pricing and payment models should be implemented.
A limited number of gene therapy medicinal products (GTMPs) have received marketing authorization (MA), of which some have been withdrawn, and even less have gained reimbursement. Many challenges that complicate GTMP market access can occur across multiple jurisdictions and decision-making contexts, but some reimbursement challenges are specific to jurisdictions. The importance of these challenges will vary according to the specific therapy being developed, the country where market access is sought, and the efforts made by developers, regulators and payers to implement solutions to overcome these barriers. This review could alert developers to challenges associated with GTMP MA and how to address them.
Introduction
Gene therapies are innovative therapies that can result in permanent improvement of patients’ lives and might, for certain diseases, even provide a cure. An overlap in the definitions of gene therapies and genetically engineered cell therapies is observed in literature 1, 2, 3. Moreover, the European regulation (EC) No 1394/200 states that products that may fall under both definitions of somatic cell therapy medicinal product (sCTMP) or tissue-engineered products (TEP) and GTMP (see Glossary) shall be considered as GTMP in Europe. Therefore, the term ‘GTMP’ is also used here to refer to all types of gene therapies and genetically engineered cell therapies (see Glossary). GTMPs aim to address the cause of a disease by correcting the genetic material in the disease-causing cells of the patient, altering the genetic material of other autologous cells to counter-act the activities of the disease-causing cells (e.g., chimeric antigen receptor T cells; CAR-T), or by providing engineered allogeneic cells to counter-act the activities of the disease-causing cells. GTMPs are mainly being developed for cancers and monogenic rare disorders [4].
As regulators became aware that existing drug assessment frameworks might not fully address the characteristics and challenges of these complex novel therapies, specific approval pathways for the regulatory evaluation of GTMPs were established. In Europe, GTMPs are classified as Advanced Therapy Medicinal Products (ATMPs), for which MA must be obtained through the centralized procedure. The evaluation of these products is performed at the European Medicines Agency (EMA) by the Committee for Advanced Therapies (CAT), which provides a draft opinion to the Committee for Medicinal Products for Human Use (CHMP). If the CHMP adopts a positive opinion, MA is granted by the European Commission (EC) [5]. In the USA, GTMPs are called Cellular and Gene Therapy Products and are regulated under the Division of Cellular and Gene Therapies (DCGT) of the Office of Tissues and Advanced Therapies (OTAT) at the Center for Biologics Evaluation and Research (CBER) of the US Food and Drug Administration (FDA) 2, 6. In addition, the Tissue and Gene Therapies Advisory Committee (CTGTAC) can provide the FDA with advice from external experts. In Canada, the Biologics and Genetic Therapies Directorate of Health Canada is responsible for the regulation of GTMPs. A ‘regulatory sandbox’ has been created in Canada for the regulatory review of advanced therapeutic products to evaluate products in collaboration with healthcare stakeholders and establish new regulatory pathways [7].
In contrast to the novel pathways that are put in place by regulators to ensure appropriate assessments of these novel therapies, most health technology assessment (HTA) bodies and payers have not adapted distinct assessment pathways for GTMPs [3]. In Europe, HTA is usually performed and payer decisions are reached at the national level by the Member States. Although efforts have been made to align assessments across European countries (e.g., EUnetHTA core model), criteria and techniques used to assess therapies and processes to ensure access still vary widely, as do payer decisions as a result. In the USA, no specific governmental institute performs centralized HTAs. US payers include ten federal government programs*, 50 state employee plans, numerous private health insurers, and employers with self-insured employer plans. These payers internally decide on reimbursement (i.e., coverage) decisions through Pharmacy and Therapeutics (P&T) committees and economic negotiations; in this process, some payers consider recommendations from the Institute for Clinical and Economic Review (ICER), which has specifically adapted its methods for these products 8, 9. All US payers have to organize reimbursement separately and coordination among private payers is prohibited by anti-trust law. Although US private health insurers are obliged to reimburse medically necessary FDA-approved therapies, including GTMPs, self-insured plans regulated under the Federal ERISA law have more freedom to define benefits and might not reimburse GTMPs. In Canada, HTA is conducted by two organizations: at the federal level by the Canadian Agency for Drugs and Technologies in Health (CADTH), and by the Institut National d’Excellence en Santé et Services Sociaux (INESSS) for the region of Quebec. By contrast, reimbursement decisions are made by the 19 different payers representing the ten provinces and three territories, and six federal programs† [10].
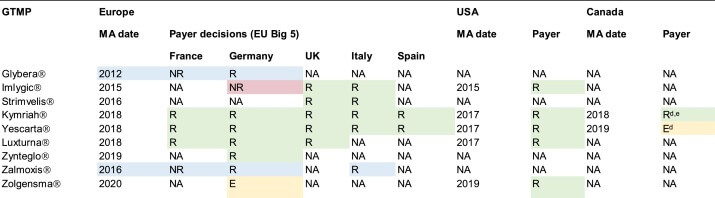
To date, nine GTMPs have received MA in Europe, five in the USA, and two in Canada (Table 1). However, these GTMPs have gained reimbursement in few countries and two (Glybera® and Zalmoxis®) have since been withdrawn from the market. Challenges to obtaining market access (covering both MA and reimbursement) and meeting postmarketing requirements to maintain conditional MA are likely to have contributed to the withdrawal of these products 11, 12. Furthermore, the limited number of authorized GTMPs and lack of reimbursement might indicate the existence of barriers. Previous research has focused on explaining some of these challenges in-depth. However, in this systematic review (the methods of which are described in File I in supplemental information online), an overview is provided of the different challenges that can be encountered when GTMP developers try to gain market access in Europe, the USA, and Canada. Moreover, we describe trends among main challenges, and potential solutions.
Table 1. Payer decisions regarding GTMPs approved in Europe, the USA, and Canadaa,b,c
Payer decisions regarding GTMPs approved in Europe, the USA, and Canada
bAbbreviations: E, reimbursement expected; MA, marketing authorization; NA, not applicable because application for marketing authorization or reimbursement has not been submitted or a decision has not yet been reached; NR, not reimbursed; R, reimbursed.
cGreen, countries where product is reimbursed; Red, countries where reimbursement of product was refused; Blue, decisions are no longer effective because product is withdrawn from the market; Yellow, recommended by HTA body but not reimbursed by any payer.
dRecommended by national HTA body on the condition of a substantial price reduction.
eReimbursed in Ontario and Quebec.
Challenges
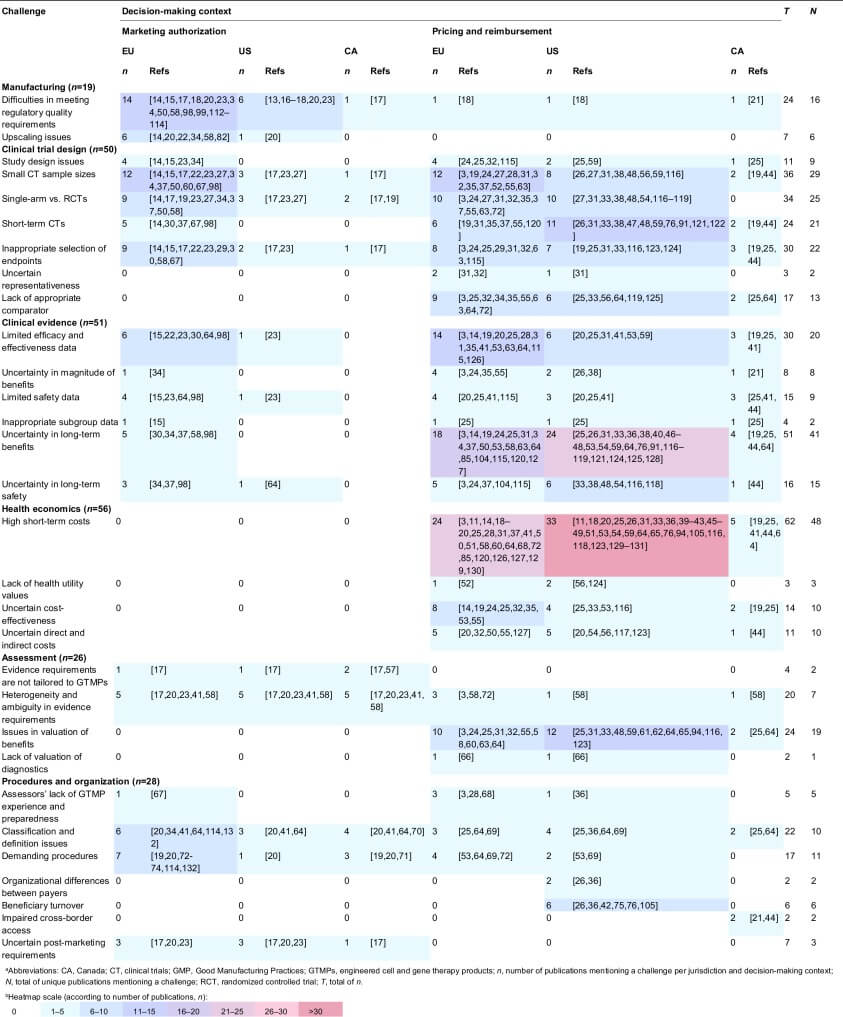
We identified 95 publications reporting on challenges in gaining market access for GTMPs (File II in supplemental information online). Data extraction from these publications resulted in the identification of 30 challenges (Table 2). For the purpose of this research, a challenge was defined as any issue originating from quality, clinical, and health economic studies and evidence, or from assessment practices, procedures, and the organization of healthcare that can affect evaluations of GTMPs by regulators, HTA bodies, and payers, or can affect developers’ efforts to obtain MA and reimbursement. Publications reported on challenges related to manufacturing (n = 19), clinical trial design (n = 50), clinical evidence (n = 51), health economics (n = 56), assessments (n = 26), and procedures and organization (n = 28). Table 2 lists the identified challenges and classifies the publications that mentioned them per jurisdiction and decision-making context. In the following section, the nature and impact of these challenges are described and case examples are provided. Subsequently, occurrence of challenges, trends and relations among these, as well as solutions are discussed.
Table 2. Heatmap of the identified GTMP market access challenges according to their frequency in the literature 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132
Table 2. Heatmap of the identified GTMP market access challenges according to their frequency in the literature
Manufacturing
Difficulties in meeting regulatory quality requirements
Cauchon et al. [13] stated ‘Aside from safety related issues that occur in the clinic, the greatest hurdle to approval is often not clinical efficacy but manufacturing/chemistry, manufacturing, and controls quality’. GTMPs are often initially developed by small-sized (academic teams or small- and medium-sized enterprises; SMEs) developers. These developers often do not prioritize manufacturing optimization early on because of a lack of experience and costs of implementing Good Manufacturing Practices (GMP). Therefore, changes to manufacturing frequently occur at late stages of development, when the GTMP is making its way toward MA applications and is facing regulatory requirements [14]. These changes result in comparability issues during regulatory evaluation [15]. Besides changes to manufacturing, novel manufacturing methods used to produce GTMPs can also challenge regulatory requirements relating to development of performance criteria, testing methodologies, and ingredient specifications 13, 16, 17. Moreover, ex vivo GTMPs using autologous cells are produced as patient-specific products, rendering the implementation of GMP even more challenging 14, 18. In an analysis of European MA applications of gene therapies, Carvalho et al. [15] identified that main quality objections reported in the MAA assessment related to issues with the production process (changes and comparability issues), drug specification, or release assay data. The additional testing needed to meet regulatory requirements also comes with additional costs; estimated at 17–45% of production costs for cell therapies [19]. Given these costs, manufacturing is often limited to a few certified manufacturing centers per country or even per continent 20, 21. Stakeholders (including developers and regulators) have been working together to resolve these issues, and the respective solutions are discussed in the ‘Solutions’ section of this review.
Upscaling issues
When GTMPs move toward late development, upscaling of production is necessary for Phase II and III clinical trials (CTs) and commercialization. Upscaling of GTMP manufacturing is difficult, especially in ex vivo autologous cell applications, where individual processes are used 14, 20. As the ‘process is the product’ for GTMPs and biologics in general, changes in manufacturing can result in changes to the product. For Glybera®, for example, changes were found to have possibly led to confounding of efficacy and safety data and the developer was obliged by EMA to repeat some clinical studies to prove comparability [22].
Clinical trial design
Study design issues
Multiple articles have discussed issues in GTMP CT design, including incompliance with Good Clinical Practices (GCP), single-center trial designs, retrospective data collection, use of historical controls, and lack of use of biomarkers 14, 15, 23, 24. In particular, single-center designs can have implications on the generalizability of outcomes because these are known to show larger effect estimates compared with multicenter studies [24]. Other frequently identified study design issues are discussed in more detail below. Carvalho et al. [15] reported that, during the assessment of clinical evidence for three of four rejected gene therapy MAs and a granted one, major GCP issues were identified; especially in trials initiated in academia, probably because of a lack of experience and resources. In a review of value assessments of six GTMPs across jurisdictions, Faulkner et al. [25] conducted an analysis of 100 HTA reports regarding transformative therapies, including GTMPs, from Australia, Canada, France, USA, and UK, to understand how HTA bodies evaluate transformative therapies. They reported that 50% of Imlygic® and Kymriah®, as well as <50% of Luxturna® HTAs noted study quality concerns.
Small clinical trial sample sizes
Gene therapy CTs are often conducted in few patients because diseases for which gene therapy are in development are often rare or ultrarare 22, 26. Although this issue was raised as a challenge in the literature, the impact on obtaining MA appears small because products have obtained MA based on evidence from small samples; potentially pointing towards flexibility of regulators in assessing GTMPs. Sample sizes of randomized controlled trials (RCTs; excluding single-arm studies) for EMA-approved ATMPs (including GTMP, sCTMP, TEP, and combined products) ranged from 99 to 512 participants [27]. In an assessment of the ATMP pipeline (including mostly Phase I and I/II trials), Hanna et al. [28] reported that almost half (47.2%) of CTs had sample sizes of <25 patients. CAR-T CTs in particular often reach very limited sample sizes (n <100) [27].
Single-arm versus randomized controlled trials
Although the golden standard for CTs is the RCT, it might not be possible or ethical to conduct RCTs for GTMPs because of a high unmet medical need, their invasiveness, and small patient populations 17, 19. In single-arm trials, historical data are often used as control, but the relative treatment effect can be overestimated if the studied population included in historical data is not matched to the CT population [24]. Abou-El-Enein et al. [27] reported that half of EMA-approved ATMPs were tested in RCTs and the other half in single-arm studies, demonstrating that the impact of this challenge on obtaining MA is also limited.
Short-term clinical trials
GTMPs have the potential to generate long-term effects; however, CTs cannot cover the full lifespan of patients and will not be able to capture whether the benefit or cure provided by the GTMP is permanent. Moreover, they are not able to identify long-term risks [26]. These long-term uncertainties are discussed in more detail under the section on clinical evidence challenges.
Inappropriate selection of endpoints
Change in endpoints over time or use of uncertain clinically relevant, nonvalidated, or surrogate endpoints has often been an issue not only in the assessment of GTMP MA applications by EMA and FDA, but also in assessments performed by HTA bodies and payers 19, 23, 29. Assessors are concerned that the use of surrogate endpoints results in larger treatment effects than when patient-relevant outcomes are used [24]. For example, EMA rejected Glybera® endpoints in their initial assessments [30]. In an analysis of GTMP HTAs, it was reported that 100% of Luxturna® and Glybera®, and 50% of Yescarta® and Kymriah® HTAs noted an uncertain link between surrogate and hard outcomes [25].
Uncertain representativeness
Given the rarity of the diseases that are targeted with GTMPs, recruitment for CTs is often difficult and participants with different baseline characteristics can be included to increase the sample size, limiting the transferability and generalizability of results [31]. In an assessment of Strimvelis® performed for the National Institute for Health and Care Excellence (NICE; British HTA body), South et al. [32] mentioned that concerns remained regarding the representativeness of the samples to the UK ADA-SCID population. Although there was a lack of clarity regarding their screening, exclusion numbers, and characteristics of the included patients, the authors concluded that the data were likely to be generalizable.
Lack of appropriate comparator
Finding an appropriate comparator for GTMPs can be difficult, especially when the therapy can lead to practical changes in clinical practice (e.g., need for additional services and facilities) or in case there are no existing treatments [33]. Although previous CT design challenges impact both regulatory evaluation and HTA, lack of an appropriate comparator mainly impacts HTA. Faulkner et al. [25] reported that 100% of Imlygic®, Yescarta® and Kymriah®, and 50% of Strimvelis® HTAs noted a lack of appropriate comparative data.
Clinical evidence
Limited efficacy and effectiveness data
Lack of demonstration of efficacy and effectiveness for GTMPs has been identified by both regulatory agencies and payers. Carvalho et al. [15] reported that, for three out of four EMA-rejected gene therapies and two out of three approved ones, there was limited evidence demonstrating efficacy [30]. Given that GTMPs are often granted conditional MA, concerns remain regarding their effectiveness at the point at which market access is sought for GTMPs. This influences the value perception by payers, who might not be willing to pay for these uncertain benefits [20]. In an analysis of GTMP HTAs, it was reported that 100% of Imlygic®, Luxturna®, Glybera® HTAs, and 50% of Strimvelis®, Yescarta® and Kymriah® HTAs noted uncertainty regarding efficacy [25].
Uncertainty in magnitude of benefits
Defining and identifying eligible, comparable patients for GTMP trials is difficult. Often patients with advanced disease stages are selected to compensate for the unknown risks of the new treatment. However, these patients can suffer irreversible damage and, therefore, the benefits they gain from GTMPs might be limited [34]. Other limitations in CT design, heterogeneity of clinical evidence, and lack of appropriate comparative data, as discussed earlier, also influence the certainty with which the magnitude of comparative benefit can be established in HTA 21, 24, 35.
Limited safety data
Limited or incomplete safety evidence has been observed in gene therapy MA applications in Europe and assessment of immunogenicity in particular is often inadequate, resulting in uncertainty regarding immunogenicity [15]. Similar to effectiveness uncertainties, often safety uncertainties also remain at the point at which market access is sought for GTMPs and influence value perception by payers [20]. Faulkner et al. [25] reported that 100% of Strimvelis® HTAs, 50% of Yescarta® and Kymriah®, and <50% of Luxturna® HTAs noted uncertainty regarding safety.
Inappropriate subgroup data
Although subgroup analysis can be valuable when assessing heterogenous clinical evidence, performing post-hoc analyses without prior specifications is not good practice. Such post-hoc analyses were reported for some gene therapies assessed by EMA, and were found to be hypothesis generating instead of confirming [15]. Although this issue does not appear to have impeded obtaining MAs, it does appear to impact assessments of payers. In an analysis of GTMP HTAs, it was reported that 100% of Glybera®, Yescarta® and Kymriah®, and 50% of Strimvelis® HTAs, as well as <50% of Luxturna® HTAs noted lack of appropriate subpopulation data [25].
Uncertainty in long-term benefits
As discussed earlier, often relatively short CTs form the basis of evaluations. Long-term benefit can be extrapolated from these trials, but limited knowledge on appropriate methods to do so results in uncertainty in long-term benefits, which was one of the major reasons why EMA initially rejected Glybera® [30]. This uncertainty also influences the value perception by payers because they are concerned that retreatment might be needed in the future 31, 36. Retreatment comes with unknown safety and efficacy, as well as considerable additional costs [3]. Faulkner et al. [25] reported that 100% of Luxturna®, Glybera®, Yescarta®, and Kymriah®, and 50% of Imlygic® HTAs noted uncertainty regarding duration of effects.
Uncertainty in long-term safety
Short CTs are also unable to address long-term safety [3]. It remains unknown how long GTMPs will remain functional in the human body and concerns exist on encountering unanticipated side effects in the years beyond duration of CTs 24, 37, 38. Therefore, GTMPs are granted conditional MAs with the requirement to monitor patients for over a decade 33, 37.
Health economics
High short-term costs
Given that GTMPs in general are developed to only require one or a limited number of administrations, high short-term costs have been quoted for them 19, 26. Examples include 1.1 million for Glybera®, which was only used for one patient in Germany, US$850 000 for Luxturna® in the USA, US$700 000 for Strimvelis®, US$125 025 for Imlygic®, US$373 000 for Yescarta®, and US$500 902 for Kymriah® 20, 25, 28, 39, 40, with prices being more elevated in the USA than the EU [41]. These high prices result in a high short-term cost per patient, which is by some perceived as an affordability threat for healthcare systems 33, 36, 42. Although one GTMP is unlikely to endanger the sustainability of healthcare budgets, it remains to be seen whether the cumulation of GTMPs could become problematic for payers 40, 43. Sustainability problems could become more prominent when GTMPs for common diseases make their way to the market [41]; although this might also provide opportunities to decrease prices [44]. It is difficult to determine a price for GTMPs because of complex logistics, the need to scale out or up, lack of transparency on initial research and development (R&D) investments and production costs, combination of fixed indirect and variable direct costs, and lack of agreement on whether prices of therapies should be value, procedure, or R&D based 18, 45, 46. The high prices also face criticism in light of efforts to reduce healthcare spending 19, 47. Some find these prices to be justified because of: (i) frequent expensive failures of other drugs in R&D; (ii) expensive R&D, manufacturing and clinical delivery of GTMPS themselves (especially for personalized ex vivo autologous types); (iii) a promise of real added value (compared with the marginal added value of me-too products); (iv) small patient populations; and (v) their innovative nature 19, 33, 41, 48, 49. Manufacturing costs of GTMPs appear to be driven by the need for highly skilled employees, ensuring regulatory requirements (GMP) are met, safety precautions (high-grade clean rooms), and lack of economies of scale for patient-specific production of ex vivo GTMPs using autologous cells 18, 19, 50, 51. Walker et al. [51] estimated the manufacturing costs of CAR-T therapies to be US$25 000–35 000 per patient.
Lack of health utility values
Given that diseases for which GTMPs are in development often are rare, have no alternative treatment options, and are underinvestigated, utility values from representative samples might not be available in the literature. In addition, because utility values have an important role in cost-effectiveness analyses, it is important to ensure that health utility in the target population of the GTMP is present or acquired to allow for accurate reimbursement decision making [52].
Uncertain cost-effectiveness
Effectiveness and, therefore, also cost-effectiveness, are uncertain at the time payers need to make a decision because: (i) GTMPs often receive MA based on limited clinical evidence for the aforementioned reasons 14, 19, 33; and (ii) lack of comparative effectiveness data [53]. Hampson et al. also stated ‘Even when products have a significant potential to confer important clinical advances over current therapies, this may not be known with a high level of certainty at the time of licensing the product. A new technology’s cost-effectiveness may be more difficult to determine in these circumstances’ [24]. An analysis of GTMP HTAs identified that 100% of Luxturna®, Yescarta®, and Kymriah®, as well as 50% of Imlygic® HTAs noted uncertainty around cost-effectiveness [25].
Uncertain direct and indirect costs
GTMP therapeutic costs do not include the ancillary medical costs of administering them, treating complications, and follow-up care. Evidence can be lacking for these costs in the intended-to-treat population 20, 54, 55, 56. In addition to these healthcare system costs, patients and their families might incur costs because they are required to travel to administration sites (Centers of Excellence), often close to a manufacturing site, and to stay there for follow-up monitoring [50].
Assessment challenges
Evidence requirements are not tailored to GTMPs
Regulators in Europe, USA, and Canada require the submission of high-quality clinical evidence, preferably obtained in RCTs. However, RCTs can often not be conducted for GTMPs as previously explained in the CT design section [17]. Although European and US regulatory frameworks for GTMPs are put in place (see Introduction), Viswanathan et al. [57] reported that there are gaps in the current Canadian regulatory framework with regards to the approval of ATMPs and that Health Canada regulators will interpret the regulations in a reasonable and flexible manner using a case-by-case approach.
Heterogeneity and ambiguity in evidence requirements
Regulatory requirements can be unclear and differ between jurisdictions, resulting in different decisions 17, 20, 23, 41. Coppens et al. described clear differences in evidence requirements for the EU and USA [23]. For example, they saw that unmet medical needs have been taken into account in EU MA decision-making, resulting in a more risk-taking approach than in the USA, although the USA has also been more risk tolerant with regards to CAR-T approvals. Papadaki et al. reported ‘Clinical development of ATMPs is also met with an inefficient assessment framework, failing to provide clear go/no-go decision criteria’ [58]. Besides differences in regulatory requirements, HTA requirements and methodologies also vary across Europe, USA, and Canada, leading to differences in access for patients [3]. Moreover, evidence requirements of regulators are often very different from those of HTA bodies and payers (e.g., regulators’ acceptance of limited clinical evidence is higher than that of payers), making it difficult to be aware of, and navigate among, all requirements [58].
Issues in valuation of benefits
Payer decision-making is generally based on value assessments. However, discussion remains over what constitutes ‘value’ in the field of health economics. Value assessment classically covers evaluations of benefit [e.g., cost-utility analysis based on quality-adjusted life year (QALYs)] and costs. GTMPs are challenging HTA practices because: (i) unique attributes that influence the value proposition are not acknowledged; (ii) evidence regarding unique promising technology attributes (e.g., long-term benefits) is not available or uncertain; and (iii) there is no clear methodological approach for these unique value scenarios, making it difficult to generate the robust health economic estimates that form the basis of any value assessment 3, 24, 25, 31, 33, 59, 60. Papadaki et al. [58] stated ‘Uncertainties around data availability and maturity question how ATMPs can meet cost-effectiveness thresholds in the existing HTA methodologies, which could disproportionally disadvantage them’. In research commissioned by NICE, Hettle et al. [24] reported that current NICE methods were likely to appropriately assess uncertainties of GTMPs, but that standard assessments might not be sufficient. Multiple authors showed that assumptions made in standard survival and benefit models resulted in the undervaluation of GTMPs, for example for CAR-T treatments and Luxturna® 55, 61, 62. In addition, standard time horizons, and cost and benefit discounting practices might need to be reconsidered for GTMPs because they are likely to come with a high upfront cost but might result in life-long benefits after a single dose 31, 32, 33, 63. Although no GTMP-specific value frameworks exist [64], it has been argued that factors beyond standard value frameworks should be considered, such as societal benefits, long-term system savings, first and only successful treatment, effect on infrastructure of care, equity, value of hope, severity of disease, value of innovation, and value of cure versus incremental benefits 3, 31, 33, 63, 65.
Lack of valuation of diagnostics
GTMPs might only provide benefits for patients with the right genetic defect or cancer type. Therefore, patients have to be accurately diagnosed. Although some diseases, such as hemophilia, can be diagnosed based on the symptoms of the patient, diagnosis for other disorders relies on genomic-based testing strategies (a form of companion diagnostics). Until now, value assessment frameworks have not allowed for a combined assessment of gene therapies and their diagnostics, but combined assessment could ensure that the opportunity cost of the gene therapy and diagnostic are quantified fully so that decision-makers are better informed [66].
Procedural and organizational challenges
Assessors’ lack of GTMP experience and preparedness
Lack of GTMP awareness, preparedness, and experience was reported for both regulators and payers. However, for regulators, this lack of experience was mostly reported in the context of the assessment by EMA of Glybera®; the first approved GTMP [67]. Later on, EMA built on that experience to update guidance [3]. With regards to HTA bodies and payers, awareness and experience is still lacking 36, 68. They are also more reluctant to acknowledge limitations of current value frameworks and to develop and adopt new methodologies in the near future [44]. Lack of appropriate tools and experience can lead to delayed assessments and uncertainty over how these products will be assessed and reimbursed 3, 68. In interviews with US payers, Barlow et al. [36] found that one-third of payers were newly aware, 40% described watchful waiting, and only 26.7% were actively involved.
Classification and definition issues
Consensus on a definition of GTMPs is lacking [69]. Moreover, classification systems differ between jurisdictions. In some jurisdictions, GTMPs are classified with other cell therapy and tissue-engineering therapies under umbrella terms as ATMPs (EU), but in other jurisdictions it is uncertain whether umbrella terms (e.g., Regenerative Medicine Advanced Therapy/RMAT, USA) cover GTMPs, or no formal regulatory definitions and classifications exist (Canada); making it difficult to know in what class a GTMP falls and what guidelines should be followed 20, 70. At the HTA level, there is a general lack of definitions and classification systems for these products [64].
Demanding procedures
According to Halioua-Haubold et al. [69], GTMPs are highly regulated under 10 US and 17 EU gene therapy-specific guidelines and regulations, making regulatory procedures demanding for GTMP developers in terms of time and resources. Procedures for GTMPs are often more complex than those for other drug classes because additional concerns need to be addressed in quality, safety, and environmental risk assessments, and exemptions, such as hospital exemptions in the EU, might apply [71]. Regulatory procedures are already demanding, but ‘companies have begun to raise the regulatory bar as a tactic to disrupt competitor companies’ according to Bubela et al. [19]. These procedures can be especially challenging for small and medium-sized enterprises (SMEs), which include many GTMP developers, which have less to no experience with these procedures, as also confirmed by EMA 17, 20, 72, 73. The first gene therapy to be approved in the EU, Glybera®, paved the regulatory pathway for GTMPs, but this required additional investments. UniQure invested an additional 15 million in Glybera® following submission to EMA [74]. Besides regulatory procedures, procedures to obtain reimbursement are also demanding and differ between EU countries and sometimes even between regions within one country 64, 72. Given that small developers might not have the necessary experience with these procedures and payer requirements, it might be difficult to obtain reimbursement [53].
Organizational differences between payers
Organizational features of payers will affect the way they will be able to mitigate the financial risks of GTMPs. In the USA, federal and state programs might be restrained as a result of annual governmental budgets and fixed premiums, national commercial payers might be able to absorb these costs well, and small payers (e.g., smaller state, regional, and self-insured employers) might need to find additional support (e.g., stop-loss insurance or reinsurance) or opt for spread payments to manage these costs 26, 36.
Beneficiary turnover
GTMPs could provide benefits over a long period of time. In a multipayer system (USA), the initial payer will invest substantial financial resources whereas, when the patient after treatment switches to another payer, a second payer will not have to make the high investment and will profit from the investments of the initial payer 26, 42, 75. For example, Medicaid has already reported a very high turnover of patients and, therefore, is likely to miss out on the benefit resulting from its GTMP investments [36]. This dynamic could disincentivize payers to invest in GTMPs [76].
Impaired cross-border access
GTMPs will likely only become available at a limited number of centers per country or even per continent because of manufacturing requirements. Cross-border (national, regional, or between insurers/formularies) access will require cross-border reimbursement systems that are currently lacking and will require agreements between treating centers, payers, and developers 21, 44.
Uncertain postmarketing requirements
Expectations from regulators can be unclear regarding postmarketing follow-up of patients, and the safety and quality measures that should be taken [20]. Moreover, these also differ between jurisdictions [17]. For example, postmarketing requirements in the EU have been related to quality, efficacy, and safety, whereas, in the USA, safety has been the main focus [23].
Occurrence, interrelation and trends
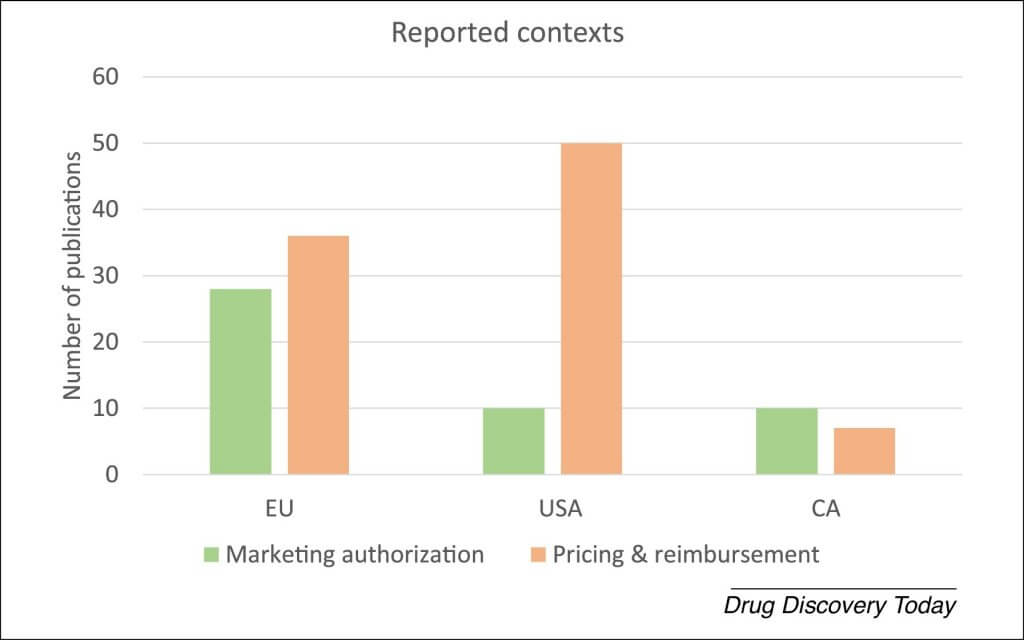
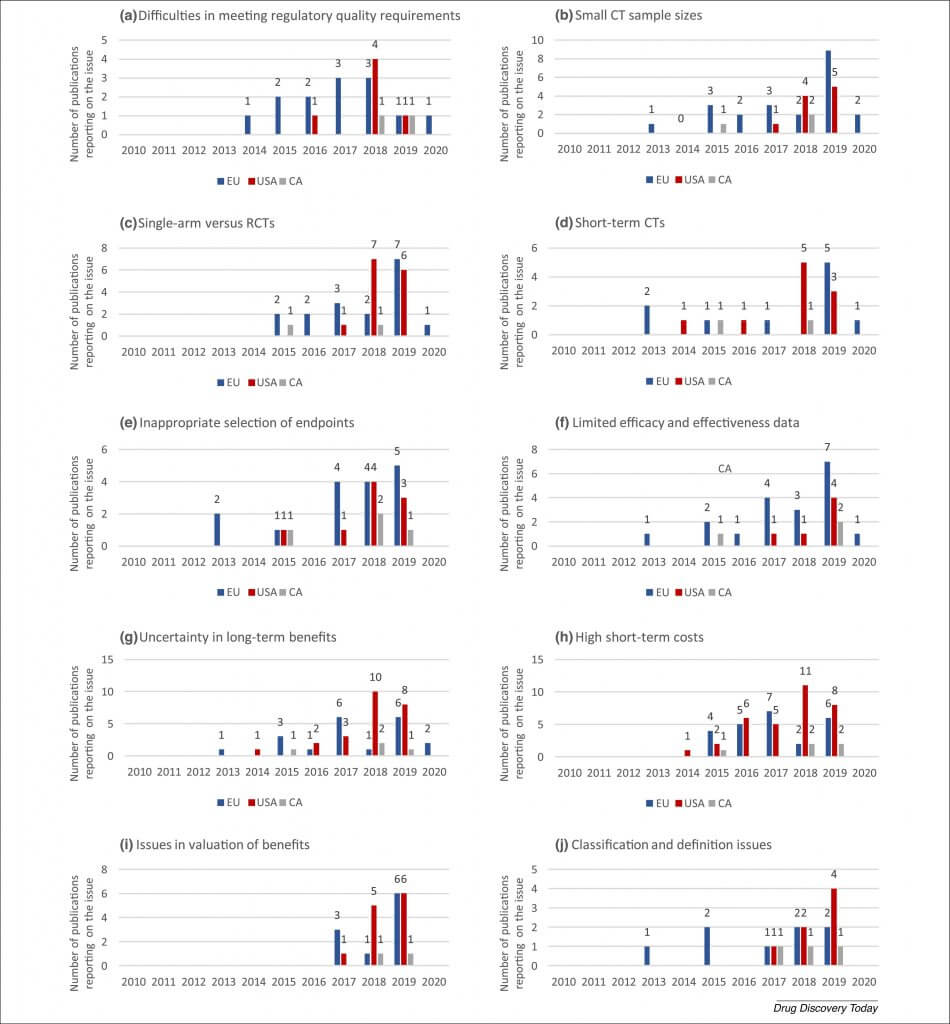
Of the publications identified in this review, 58% reported on challenges in the USA, 56% in the EU, and 14% in Canada. EU and Canadian publications covered MA and pricing and reimbursement phases almost equally, whereas US publications mainly focused on pricing and reimbursement issues, potentially indicating that these have a more prominent role in market access in the USA (Fig. 1).
Figure 1. Contexts that identified publications reported on. Abbreviations: CA, Canada; CT, controlled trial; EU, European Union; GTMP, gene therapy medicinal products; MA, marketing authorization; RCT, randomized controlled trial; PR, pricing and reimbursement. Interpretation of the literature by authors.
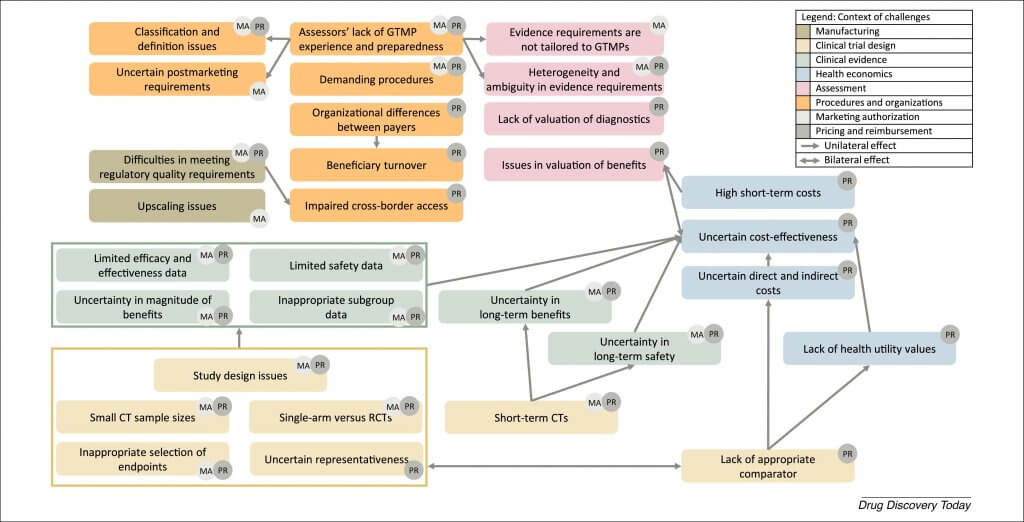
Publications mostly reported on challenges in pricing and reimbursement of GTMPs (77%) and less on challenges in obtaining MA (35%). From Table 2 and the description of challenges, several trends can be observed. Although many challenges were mentioned across multiple jurisdictions and decision-making contexts, certain challenges appear to be specific to legislations or decision-making contexts. For example, quality (manufacturing) issues were specific to the MA phase. As can be expected, health economic and procedural reimbursement issues were specific to the pricing and reimbursement phase, with issues caused by a multipayer system being specific for the USA and cross-border access being a specific challenge for Canada. Although not retrieved in the literature, ‘impaired cross-border access’ might also be present in the other jurisdictions, because ‘Difficulties in meeting regulatory quality requirements’ results in limited availability of administration sites and this challenge has also been identified for Europe and the USA. We qualitatively identified that challenges also appear to be highly interlinked, and observed that a certain domino effect can be observed because challenges affect each other (Fig. 2). For example, CT design issues affect clinical and health economic issues, and multiple issues also appear to be caused at least partially by the lack of experience of developers. The existence of this domino effect also means that challenges can be overcome and even prevented when early root challenges are addressed (e.g., CT design challenges).
Figure 2. Relation between challenges and the contexts in which they can occur.
The ten most-frequently identified challenges were ‘Difficulties in meeting regulatory quality requirements’, ‘Small CT sample sizes’, ‘Single-arm versus RCTs’, ‘Short-term CTs’, ‘Inappropriate selection of endpoints’, ‘Limited efficacy and effectiveness data’, ‘Uncertainty in long-term benefits’, ‘High short-term costs’, ‘Issues in valuation of benefits’, and ‘Classification and definition issues’ (T scores in Table 2). All issues, except for ‘High short-term costs’, were first reported in the EU context and later in other jurisdictions (Fig. 3), confirming expectations, given that EU regulators were the first to approve a GTMP (Glybera®). Reporting on six of these nine challenges started in the EU in 2013 (following the European MA of Glybera® in 2012), and, on average, reporting in the USA and Canada was delayed by 2 and 2.1 years, respectively. However, reporting on single-arm and short-term CT issues peaked 1 year earlier in the USA (2018) than in the EU (2019). Reporting on issues in valuation of benefits started most recently (2017). ‘Difficulties in meeting regulatory quality requirements’ is the only challenge on which reporting has started to decrease over time (n = 8 in 2015, n = 3 in 2019, 62.5% reduction); potentially reflecting that developers are increasingly able to overcome this hurdle. Carvalho et al. [15] also reported ‘In general, there has been a clear trend regarding quality data acceptability by CAT/CHMP. This could either be a result of the increased experience of regulators with GTMP assessment or the submission of more robust quality data by the applicants’. The solutions that enabled developers to overcome these barriers are discussed in the later ‘Solutions’ section. Reporting is still increasing over time for ‘Small CT sample sizes’ (n = 4 in 2017, n = 14 in 2019, 250% increase), ‘Single-arm versus RCTs’ (n = 4 in 2017, n = 13 in 2019, 225% increase), ‘Short-term CTs’ (n = 1 in 2017, n = 8 in 2019, 700% increase), ‘Inappropriate selection of endpoints’ (n = 5 in 2017, n = 8 in 2019, 60% increase), ‘Limited efficacy and effectiveness data’ (n = 5 in 2017, n = 13 in 2019, 160% increase), ‘Uncertainty in long-term benefits’ (n = 9 in 2017, n = 15 in 2019, 67% increase), ‘High short-term costs’ (n = 12 in 2017, n = 16 in 2019, 33% increase), ‘Issues in valuation of benefits’ (n = 4 in 2017, n = 13 in 2019, 225% increase), and ‘Classification and definition issues’ (n = 3 in 2017, n = 7 in 2019, 133% increase).
Figure 3. Evolution of the most-frequently mentioned issues in the literature over time. The frequency presented reflects the total of unique publications per year that mentioned a challenge in a specific jurisdiction. Challenges include: (a) difficulties in meeting regulatory quality requirements; (b) small clinical trial (CT) sample sizes; (c) single-arm versus randomized CTs (RCTs); (d) short-term CTs; (e) inappropriate selection of endpoints; (f) limited efficacy and effectiveness data; (g) uncertainty in long-term benefits; (h) high short-term costs; (i) issues in valuation of benefits; and (j) classification and definition of issues. Abbreviations: CA, Canada; EU, European Union. *Medicare, Medicaid, State Children’s Health Insurance Program, Children and Youth with Special Health Care Needs, Tricare, Veterans Health Administration, Indian Health Service, Federal Employee Health Benefits, Substance Abuse and Mental Health Services Administration, and Refugee Health Promotion. †First Nations people living on reserves, Inuit people, serving members of the Canadian Armed Forces, eligible veterans, inmates in federal penitentiaries, and some groups of refugee claimants. No federal health programs exist for non-status First Nation and Métis people.
Similarities in challenges were observed between GTMPs and other drug classes, covering biologics, orphan drugs, vaccines, and medical devices and surgeries. GTMPs can be categorized as biological therapies, which otherwise include therapies based on monoclonal antibodies, enzymes, and hormones. As for all biological therapies ‘the process is the product’ applies, for other non-GTMP biologics, similar manufacturing challenges have been reported. In addition, CT design (including GCP and selection of endpoints) and clinical evidence (e.g., proof of efficacy, long-term efficacy and safety, and post-hoc analyses) challenges have been shown to occur during the MA phase of other biologics [77]. Besides GTMPs falling under the biologics umbrella, half of the approved GTMPs have also received orphan designation 23, 60. Given that GTMPs and other orphan drugs target small patient populations with often limited available alternatives and high unmet medical needs, similar challenges can be observed in terms of CT sample sizes, single-arm versus. RCTs, selection of endpoints, representativeness, comparators, and amount of efficacy and safety evidence 78, 79, 80, 81. Moreover, it has been reported that traditional HTA processes use methods that also do not translate well to the assessment of these other orphan drugs [60]. Another two challenges that are nonspecific to GTMPs are the valuation of life-long benefits following an upfront cost and the beneficiary turnover challenge because these are also dealt with for vaccines, as well as medical devices and surgeries. Thus, many of the challenges identified in this review are not new or specific to GTMPs. However, it appears that the avant-garde nature, magnitude of uncertainties, prices of GTMPs, and the co-occurrence of challenges work as a magnifying glass for these challenges and their impact on healthcare systems.
Solutions
Although many challenges were identified in the literature, potential solutions were also suggested.
Development of quality standards, manufacturing facilities, and upscaling solutions
To ensure manufacturing meets regulatory needs, standards are in development [16]. The EMA is proposing quality data certification (only for SMEs) and quality data assessment via scientific advice to identify any quality issues before MA applications [15]. Moreover, support platforms, such as the Cell and Gene Therapy Catapult (UK), Centre for Commercialization of Regenerative Medicine (CCRM; Canada), and CellCAN (Canada), are working on the establishment of new GMP processes for GTMPs [19]. New machines that can count, separate, and manipulate cells, and new bioreactors, such as the iCELLis Nano, which meet regulatory requirements and can ensure safe upscaling are also being developed 20, 82. Regarding manufacturing issues of ex vivo autologous GTMPs, efforts are made to investigate the use of allogeneic cells to create universal GTMPs. This could reduce manufacturing costs and lead to process improvements. In a model by Harrison et al. [83], it was shown that a reduction from US$495 780 to US$44 460 per dose could be realized by switching from autologous to allogenic production of CAR T-cells.
Addressing clinical uncertainties
As discussed earlier, it might not be possible to conduct RCTs for GTMPs. Therefore, clinical evidence might need to be strengthened with additional data from systematic reviews, meta-analyses of studies and registries, and evidence mapping for heterogeneous data [27]. However, the added value of these data depends on their timely presence and the quality of the clinical studies and their reporting. To ensure correct reporting of GTMP CTs, Abou-El-Enein et al. [27] created a checklist of questions to be addressed.
Regulators have shown flexibility in granting GTMPs conditional MA to allow for timely access while uncertainties can be addressed in postmarketing studies and assessed in periodic reports and reassessments 15, 30, 84. However, at the HTA and payer level, reimbursement appears to be mainly affected by the limited availability of evidence [19]. Therefore, more collaboration is needed among regulators, HTA, payers, and industry across jurisdictions to manage uncertainty while providing early access to these potential high-value therapies. Although accelerated and conditional approval pathways are available and should be taken advantage of, misuse of rare disease populations with high unmet medical needs to justify value and limited data should be prevented [20].
With the application of conditional MA and reimbursement, the need for infrastructures that can collect postmarketing real-world evidence (RWE) is high 15, 19, 33. The set-up of RWE infrastructure also comes with its own challenges and additional costs 37, 85. Use of existing registries owned by neutral accessible international bodies could provide a solution, as suggested by the World Haemophilia Federation, of which the WFH World Bleeding Disorders Registry (WBDR) could be used for hemophilia GTMPs [86]. In a review by Jorgensen et al. [37], 58 cell or gene therapies registries, spanning 47 indications, were identified in the UK. Oncology appears to be especially well placed for RWE collection because many registries already exist and can be updated to meet conditional reimbursement requirements. In the UK, two-thirds of non-oncology indications have registries in place, but almost none meet conditional reimbursement requirements [37]. Limited resources endanger data entry compliance and Kefalas et al. [87] reported that, for CAR-Ts with conditional reimbursement, the administrative burden results in a 4.6-fold increase in administrative resource use.
Towards new price setting procedures, value frameworks and payment models
As discussed earlier, there is a lack of agreement on whether price setting should be value, procedure, and/or R&D based. Although value-based pricing has increasingly received attention and been implemented in healthcare systems, value-based pricing might not result in price reductions. Value-based pricing for GTMPs might still result in high prices if they are based on savings that GTMP could generate over a lifetime. To account for this, prices can be corrected by reducing the timespan used to calculate a value-based price to the life expectancy with standard therapy, setting willingness-to-pay thresholds per gained QALY, setting QALY price caps, and implementing shared saving measures [59]. If prices are based on R&D and manufacturing costs, more insight is needed on those exact costs. To this end, key cost drivers in manufacturing have been identified, leading to the proposal of new cost models 18, 88. Generally speaking, more transparency is needed on R&D investments and price setting to generate public trust [49].
New value frameworks are developed to ensure that GTMPs are correctly valued. For example, ICER updated their methods for GTMPs and orphan drugs in general, and the International Society for Pharmacoeconomics and Outcomes Research (ISPOR) released a (non-GTMP specific) comprehensive value ‘flower’ framework 9, 89. By contrast, some payers, such as NICE, believe that current assessment frameworks can flexibly adapt to these therapies. The rise of GTMPs and their market access challenges provide the opportunity for stakeholders to seek consensus on what elements contribute to value of therapies and how new elements can be considered in decision-making [33]. New elements have been discussed before and their importance could be quantified by eliciting patient preferences for elements that go beyond health gain in patient preference studies 63, 90. A comprehensive approach could then be taken to take into account survival, quality of life, effectiveness, safety, patient perspective, and cost evidence simultaneously, higher cost-effectiveness threshold for therapies that provide value beyond the QALY could be set, or elements could be integrated in novel quantitative decision-making methodologies, such as multicriteria decision analysis (MCDA) 26, 65. By contrast, standard cost-effectiveness modeling can also be adapted to account for the unique features of GTMPs. Analyses can be added to quantify the importance of uncertainties and whether it is worth gathering additional evidence to reduce uncertainties, including Value of Information (VoI) modeling, scenario analysis, sensitivity analysis, and cost-effectiveness acceptability curves 19, 24, 63, 91. Drummond et al. [31] created a checklist to determine which aspects of economic evaluations should be modified based on characteristics of GTMPs. Although new methodologies might be able to resolve valuation issues of GTMPs, changes to and harmonization of, current HTA processes could be needed to allow for their use, raising concerns about comparability with past assessments [63].
Given that GTMPs come with unprecedented high prices and clinical uncertainties, several reimbursement models have been proposed, including managed-entry agreements (MEA), new financing mechanisms, and US-specific solutions that address multipayer challenges (Table 3). These models also have their own challenges, such as laws and regulations that prevent implementation or the need for RWE generation. More research is needed to optimize their use 19, 26, 36. Models can also be combined, for example, pay-for-performance with spread payments (performance-based annuities), to address uncertainties while also managing budget impact 14, 47, 92. Pay for performance agreements have already been established for Luxturna®, Strimvelis®, and Kymriah® 39, 76, 93, 94. In interviews with US payers, Barlow et al. reported that 47% of payers support new payment models, especially performance-based agreements and risk pooling [36]. Besides implementation of new payment models, investments should also be made in diagnosis so that appropriate patients can be identified and resources used wisely 26, 36. It might also be possible that a generation of ‘biosimilar-GTMPs’ will reduce prices once patents and other exclusivity rights of the originators expire. However, it remains unknown whether current regulations for biosimilars of originator biologicals (protein-based) would also apply to ‘biosimilar-GTMPs’ or how they would otherwise be regulated [95].
Table 3. Novel payment models
Class
Principle
Type
Description
Managed-entry agreements
Outcome-based agreements
Pay for performance (i.e., rebates, payment-by-result, money-back, risk-sharing, or mile-stone based) 3, 14, 26, 36, 48, 91, 92
Refunds can be given to payers for nonresponsive patients or payers only pay costs for responsive patients.
Access with evidence development (i.e., coverage with evidence development)
Reimbursement can be granted on condition that additional evidence is generated
Financial-based agreements
Price-volume agreements [3]
Price can be reduced when sales volume exceeds a threshold
Price discounts (i.e., price control) [3]
Reductions can be given under specific conditions (confidential and does not affect the list price)
Cost-sharing [3]
Discount for one or multiple cycles of treatment for certain patients
One payment per treated patient, regardless of number of services provided
Payer risk reduction
Stop-loss insurance 26, 36
Payers get reimbursed if expenditure for treatment crosses certain threshold
Reinsurance 26, 36, 92
Insurance policy protects payer from financial risk
Risk pools 3, 92
Expenditures on high-cost patients are pooled across multiple payers
Healthcoin 3, 42, 105
Care currency, exchangeable for US dollars, used to pay initial payer for beneficiary who is switching to another payer
Integration in plan design and premiums [36]
Costs can be integrated in premiums of healthcare plans, leading to premium escalations
Support, stakeholder interaction and partnering
Given a lack of experience, and demanding procedures and requirements, it is crucial that developers get support in their translational activities and interact with regulators, HTA bodies, and payers 33, 96. Different support platforms are available to GTMP developers, such as the Cell and Gene Therapy Catapult in the UK and the Centre for Commercialization of Regenerative Medicine (CCRM) in Canada (Table 4) 19, 97. In addition, regulators have set up different support programs to allow for interaction with, and incentivize, developers 5, 20, 44, 50. Early scientific advice can be provided in parallel by FDA and EMA [19], but increased harmonization between regulators, for example, through the ICH, could further incentivize developers and facilitate the conduct of international CTs and commercialization [17]. These programs could, in combination with conditional MA, and MA under exceptional circumstances, accelerate GTMP market access. Although these programs exist, developers do not often use assistance available from regulators [15].
Table 4. Support platforms and programs for GTMP developers
Organization/program
Support
Europe
EMA 5, 44
Innovation Task Force (ITF)
Scientific, legal, and regulatory advice
Small and medium-sized enterprise (SME) office
Fee reductions and exemptions
Direct assistance on regulatory procedures
Assistance with translations of product information
Guidance on clinical data publication
Workshops and trainings
Orphan designation
Protocol assistance
Tailored scientific advice
Market exclusivity (10 years or even 12 years if pediatric investigation plan is agreed upon)
Fee reductions (in some cases)
Priority medicines (PRIME) schedule
Early access procedures and accelerated assessment
Combination with conditional marketing authorization or compassionate use possible
Accelerated assessment
Reduces CHMP timeframe to 150 days
UK
Cell and Gene Therapy Catapult 19, 97
Industrialization
Manufacturing
Regulatory affairs
Health economics and market access
Nonclinical safety
Clinical operations
USA
FDA [44]
Orphan Product designation
Development incentives (e.g., tax credits for qualified clinical testing)
Exemption from drug user fees
Fast Track
More frequent meetings with FDA on drug development plan and data collection
More frequent written communication from FDA on clinical trial design and use of biomarkers
Eligibility for Accelerated Approval and Priority Review, if relevant criteria met
Rolling review
Breakthrough therapy
All fast track designation features
Guidance on drug development program from Phase I
Organizational commitment
Accelerated approval
Approval based on surrogate or intermediate clinical endpoint
Priority review
Reduced review duration (6 instead of 10 months)
Rare Pediatric Disease Priority Review
Grants voucher for priority review of subsequent marketing application for different product
Canada
Health Canada [44]
Priority Review
Shorter review time-frames
Notice of Compliance with Conditions
Potential shorter time to approval and market
Enhanced post-market surveillance initiatives
Centre for Commercialization of Regenerative Medicine (CCRM) [19]
Regulatory affairs
Technology review
Market assessment
Competitive landscape analysis
Patent strategy
Commercial path support
Manufacturing facilities and support
Early interaction with both regulators and HTA (Table 4) has been found to be especially beneficial because it could allow for appropriate CT designs, best use of small populations, agreement on validation and use of surrogate endpoints, agreement on comparators, appropriate statistical analysis, and agreement on postlaunch evidence gathering 3, 5, 33, 50. The cases of Strimvelis® and Zynteglo® in the EU prove that interaction between developers, regulators, HTA bodies, and patients during development can increase the chance of success 98, 99. Another example of multistakeholder interaction is the hemophilia GTMP pipeline, where products are moving toward MA phases, and a core outcomes set (coreHEM) was created with patients, clinicians, researchers, regulators, public funding agencies, HTA assessors, payers and drug developers with the aim to ensure that all gene therapy trials in hemophilia report on the same outcomes and can be compared 29, 86. Forming partnership among developers and other stakeholders, while maximizing the use of flexible licensing and reimbursement tools, can facilitate GTMP market access 58, 99. Therefore, new collaboration platforms, such as specialized manufacturing and delivery platforms, could be created through public–private partnerships [58].
Patient involvement has also been found to have pushed GTMP development and access forward. Given that, in many cases, GTMPs are developed for rare diseases and knowledge on these diseases is limited, patients can contribute to development and assessment through their experience on living with these diseases. Patient organizations have participated in the development of EU legislation, are represented in the CAT, and have advocated for affordability and accessibility of GTMPs 100, 101, 102. Patient engagement could address bias in research and scientific misconduct, raise awareness and support for CTs, and enhance long-term follow-up by co-creating registries with patient organizations, ensuring adherence to follow-up appointments and tests, and supporting interpretation of results to understand what improvements mean to patients 26, 100, 102. Trust, transparency, and patient ownership of data could promote patient engagement even further [102]. Increasingly, patients’ perspectives and preferences are also sought and considered in weighing of benefits and risks, defining acceptability of risks and benefits, and establishing value propositions for MA and reimbursement applications [100]. Especially for innovative products, listening to the patient perspective is crucial [100]. To engage patients in individual treatment decisions on GTMPs, coordination, coaching and clear communication on treatment expectations, potential long-term consequences, costs, reimbursement, and responsibilities are needed 26, 38.
Future perspectives
As stakeholders are gaining experience with GTMPs and novel guidelines and models are being developed, collaboration across stakeholders and jurisdictions could be stimulated to ensure equality in access to GTMPs. Moreover, cross-border access should be arranged. Here, partnering with different stakeholders is crucial.
As discussed earlier, increased harmonization among regulator and payer evaluation frameworks could reduce developer uncertainty and increase efficiency for all. Cross-jurisdictions and cross-regulatory/payer early advice would be the ideal way to reach agreements and incentivize developers to pursue reimbursement in all involved countries. Educational workshops for developers could also help familiarize them with the needs of regulators and payers. To further understand what differences exist in challenges between jurisdictions, an analysis of assessment reports among regulators (EMA, FDA, and Health Canada) and among payers of the same GTMPs could be conducted. Moreover, the ten most-frequently identified challenges (‘Difficulties in meeting regulatory quality requirements’, ‘Small CT sample sizes’, ‘Single-arm vs. RCTs’, ‘Short-term CTs’, ‘Inappropriate selection of endpoints’, ‘Limited efficacy and effectiveness data’, ‘Uncertainty in long-term benefits’, ‘High short-term costs’, ‘Issues in valuation of benefits’, and ‘Classification and definition issues’) form key areas for future research because solutions for these challenges could significantly improve GTMP market access.
Arguably, GTMPs form the current pinnacle of medicine and have the potential to change patients’ lives dramatically. Therefore, we should find ways to bring them to the patient. In doing so, attention should be given to increasing public trust in healthcare decision making and pricing [60]. Transparency here is key. The interlinkage of price, value, and payment models makes it difficult to identify where adjustments should be made to guarantee correct assessment and access to GTMPs 33, 94. Therefore, integrated solutions could be developed that, for example, only correct uncertainties in one of these three related areas to prevent double-punishment.
When more GTMPs become available, patients and society might increasingly face access difficulties related to the location of delivery centers. It might be necessary to centralize diagnostic processes, delivery, and real-world follow-up in centers of excellence [33]. To enhance the role of RWE in continued market access of GTMPs, it would be ideal to develop RWE infrastructure and requirements on an international or even global level [3].
To propel orphan drug and GTMP development forward, great efforts have been made by developers and regulators, as well as patients with rare diseases and their families. However, healthcare systems are now trying to limit expenses for rare diseases [60]. In addition, the currently most-reimbursed GTMPs, most regenerative medicines in development, and most orphan-designated drugs target oncology indications 43, 103, 104. If current orphan drug incentives and GTMP-access solutions in the future are found to adversely discriminate against monogenic diseases, orphan drug acts might require reform to restore equity.
Concluding remarks
GTMPs are a heterogeneous class of medicines with which experience is limited to date. This systematic literature review catalogued challenges that could inhibit market access of GTMPs in the EU, USA, and Canada. The often interrelated challenges combine GTMP unique challenges as well as challenges that apply to broader categories, such as biologics and orphan drugs. The importance of specific challenges and solutions identified in this review will vary according to the therapy being developed, and the country where market access is sought.
Challenges can be turned into opportunities for individual and collaborative action. This work suggests that HTAs, payers, regulators, patients, and developers should: (i) seek support and early joint interactions to come to agreements on CT design, manufacturing, and the valuation of benefits; (ii) discuss conditional MA and reimbursement mechanisms that allow for postauthorization follow-up of long-term efficacy and safety; (iii) develop RWE infrastructure and requirements on an international or even global level; and (iv) explore innovative pricing and payment models that can be implemented efficiently.
Author contributions
E.v.O. and S.M. designed the search query that was subsequently approved by M.T., H.S., M.T,. E.M., I.H., and S.S.. All authors contributed to the identification of gray literature and provided insights into the regulatory and market access processes in their respective country. E.v.O. and S.M. performed title/abstract and full-text screening of the retrieved articles and selected articles to be included in the review. E.v.O. extracted data from the included articles and produced the first draft of the manuscript, which was subsequently revised and finalized with all authors. All authors approved the final manuscript.
Declaration of interest
S.S. provided advice to Novartis about the design of a managed entry agreement for an advanced therapy medicinal product.
Acknowledgements
The authors would like to thank all members of the ExACT project, Eric Meslin, and all Council of Canadian Academies colleagues for their support of this study and E.v.O.’s secondment. Special thanks to Anu Shukla-Jones, Michelle Mujoomdar, Matthew Lucas, Chris McMaster, Etienne Richer, Susan Zimmerman, Craig Hasilo, Hanns Lochmuller, Ma’n H. Zawati and team, Siofradh McMahon, Durhane Wong-Rieger, Maureen Smith, Danika Goosney, Cindy Bell, Karen Dewar, and Cate Murray and team for sharing their insights on the Canadian policy, research, and cell and gene therapy ecosystem during the secondment. No external funding was used for the conduct of this study, but learnings gained through a secondment of E.v.O at the Council of Canadian Academies were used to inform the study design and the discussion of the manuscript. This secondment was financed by the ‘European network staff eXchange for integrating precision health in the health Care Systems’ (ExACT) project. The ExACT project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 823995. The views expressed in this paper are the authors’ and may not reflect those of the ExACT project, the Horizon 2020 programme or the Council of Canadian Academies.
References
A. Ridgway, et al. Regulatory oversight of cell and gene therapy products in Canada M.C. Galli, M. Serabian (Eds.), Regulatory Aspects of Gene Therapy and Cell Therapy Products: A Global Perspective, Springer International Publishing (2015), pp. 49-71
K. Elverum, M. Whitman Delivering cellular and gene therapies to patients: solutions for realizing the potential of the next generation of medicine Gene Ther. (2019) https://www.nature.com/articles/s41434-019-0074-7
Alliance for Regenerative Medicine Getting Ready: Recommendations for Timely Access to Advanced Therapy Medicinal Products (ATMPs) in Europe Alliance for Regenerative Medicine (2019)
S.R.P. Kumar, et al. Clinical development of gene therapy: results and lessons from recent successes Mol. Ther. – Methods Clin. Dev., 3 (2016), p. 16034
G. Detela, A. Lodge EU regulatory pathways for ATMPs: standard, accelerated and adaptive pathways to marketing authorisation Mol. Ther. Methods Clin. Dev., 13 (2019), pp. 205-232
M. Mendicino, et al. Current state of U.S. Food and Drug Administration regulation for cellular and gene therapy products: potential cures on the horizon Cytotherapy, 21 (2019), pp. 699-724
Health Canada Agile Regulations for Advanced Therapeutic Products and Clinical Trials Health Canada (2019)
Institute for Clinical and Economic Review Guide to Understanding Health Technology Assessment (HTA) Institute for Clinical and Economic Review (2018)
Institute for Clinical and Economic Review Adapted Value Assessment Methods for High-Impact ‘Single and Short-Term Therapies’ (SSTs) Institute for Clinical and Economic Review (2019)
Alberta Health Services (XXXX) Misconception: All Indigenous People Get Free Health Care, Alberta Health Services.
M. Senior After Glybera’s withdrawal, what’s next for gene therapy? Nat. Biotechnol., 35 (2017), pp. 491-492
Anon Uniqure withdraws 1m drug Glybera from market European Biotechnology (2017) April 21
N.S. Cauchon, et al. Innovation in chemistry, manufacturing, and controls-a regulatory perspective from industry J. Pharm. Sci., 108 (2019), pp. 2207-2237
M.S. Corbett, et al. Innovative regenerative medicines in the EU: a better future in evidence? BMC Med., 15 (2017), p. 49
M. Carvalho, et al.Hurdles in gene therapy regulatory approval: a retrospective analysis of European Marketing Authorization Applications Drug Discov. Today., 24 (2019), pp. 823-828
J.A. Arcidiacono, et al. FDA and NIST collaboration on standards development activities supporting innovation and translation of regenerative medicine products Cytotherapy, 20 (2018), pp. 779-784
D.G.M. Coppens, et al. Global regulatory differences for gene- and cell-based therapies: consequences and implications for patient access and therapeutic innovation Clin. Pharmacol. Ther., 103 (2018), pp. 120-127
M. Abou-El-Enein, et al. Putting a price tag on novel autologous cellular therapies Cytotherapy, 18 (2016), pp. 1056-1061
T. Bubela, et al. Bringing regenerative medicines to the clinic: the future for regulation and reimbursement Regen. Med., 10 (2015), pp. 897-911
E. Atilla, et al. Cellular therapies: Day by day, all the way Transfus. Apher. Sci., 57 (2018), pp. 187-196
Canadian Agency for Drugs and Technologies in Health Tisagenlecleucel for Acute Lymphoblastic Leukemia and Diffuse Large B-cell Lymphoma: Recommendation Canadian Agency for Drugs and Technologies in Health (2019)
T.R. Cheever, et al. Perspectives on best practices for gene therapy programs Hum. Gene Ther., 26 (2015), pp. 127-133
D.G.M. Coppens, et al. A decade of marketing approval of gene and cell-based therapies in the United States, European Union and Japan: An evaluation of regulatory decision-making Cytotherapy, 20 (2018), pp. 769-778
R. Hettle, et al. The assessment and appraisal of regenerative medicines and cell therapy products: an exploration of methods for review, economic evaluation and appraisal Health Technol. Assess., 21 (2017), pp. 1-204
E. Faulkner, et al. Are global health systems ready for transformative therapies? Value Health., 22 (2019), pp. 627-641
Anon AMCP Partnership Forum: designing benefits and payment models for innovative high-investment medications J. Manag. Care Spec. Pharm., 25 (2019), pp. 156-162
M. Abou-El-Enein, S.P. Hey Cell and gene therapy trials: are we facing an ‘evidence crisis’? EClinicalMedicine, 7 (2019), pp. 13-14
E. Hanna, et al. Advanced therapy medicinal products: current and future perspectives J Mark Access Health Policy., 4 (2016)
A. Iorio, et al. Core outcome set for gene therapy in haemophilia: Results of the coreHEM multistakeholder project Haemophilia, 24 (2018), pp. e167-e172
L.M. Bryant, et al. Lessons learned from the clinical development and market authorization of Glybera Hum. Gene Ther. Clin. Dev., 24 (2013), pp. 55-64
M.F. Drummond, et al. Analytic considerations in applying a general economic evaluation reference case to gene therapy Value Health., 22 (2019), pp. 661-668
E. South, et al. Strimvelis((R)) for treating severe combined immunodeficiency caused by adenosine deaminase deficiency: an evidence review group perspective of a NICE Highly Specialised Technology Evaluation Pharmacoecon. Open., 3 (2019), pp. 151-161
G. Hampson, et al. Gene therapy: evidence, value and affordability in the US health care system J. Comp. Eff. Res., 7 (2018), pp. 15-28
N.M. Mount, et al. Cell-based therapy technology classifications and translational challenges Philos. Trans. R. Soc. Lond. B Biol. Sci., 370 (2015)
National Institute for Health and Care Excellence Tisagenlecleucel for Treating Relapsed or Refractory Diffuse Large B-Cell Lymphoma after 2 or More Systemic Therapies NICE (2019)
J.F. Barlow, et al. Are payers ready, willing, and able to provide access to new durable gene therapies? Value Health., 22 (2019), pp. 642-647
J. Jorgensen, et al. Data collection infrastructure for patient outcomes in the UK – opportunities and challenges for cell and gene therapies launching J. Mark Access Health Policy, 7 (2019)
Institute for Clinical and Economic Review A Look at CAR-T Therapies Institute for Clinical and Economic Review (2018)
G. Hampson, et al. Gene therapy: evidence, value and affordability in the US health care system J. Comp. Eff. Res., 7 (2018), pp. 15-28
T. Morrow Novartis’s Kymriah: harnessing immune system comes with worry about reining in costs Manag. Care., 26 (2017), pp. 28-30
V. Shukla, et al. The landscape of cellular and gene therapy products: authorization, discontinuations, and cost Hum. Gene Ther. Clin. Dev., 30 (2019), pp. 102-113
A. Basu, et al. Financing a cure for diabetes in a multipayer environment Value Health, 19 (2016), pp. 861-868
C. Quinn, et al. Estimating the clinical pipeline of cell and gene therapies and their potential economic impact on the US healthcare system Value Health, 22 (2019), pp. 621-626
A. Sinclair, et al. Gene Therapy: An Overview of Approved and Pipeline Technologies CADTH Issues in Emerging Health Technologies (2016)
W.F. Kaemmerer How will the field of gene therapy survive its success? Bioeng. Transl. Med., 3 (2018), pp. 166-177
S.H. Orkin, P. Reilly Paying for future success in gene therapy Science, 352 (2016), pp. 1059-1061
T.A. Brennan, J.M. Wilson The special case of gene therapy pricing Nat. Biotechnol., 32 (2014), pp. 874-876
M. Senior Rollout of high-priced cell and gene therapies forces payer rethink Nat. Biotechnol., 36 (2018), pp. 291-292
F. Xie Highly priced gene therapies: a wake-up call for early price regulation Pharmacoeconomics, 36 (2018), pp. 883-888
G. Narayanan Translation and reimbursement: the twin challenges for cell and gene therapies reflections of an ex-regulator Hum. Gene Ther. Clin. Dev., 27 (2016), pp. 93-95
A. Walker, R. Johnson Commercialization of cellular immunotherapies for cancer Biochem. Soc. Trans., 44 (2016), pp. 329-332
C.L. Halioua-Haubold, et al. Potential lifetime quality of life benefits of choroideremia gene therapy: projections from a clinically informed decision model Eye (Lond), 33 (2019), pp. 1215-1223
D. Driscoll, et al. The high cost of high tech medicine: planning ahead for market access Stem Cells Transl. Med., 6 (2017), pp. 1723-1729
T. Jain, M.R. Litzow No free rides: management of toxicities of novel immunotherapies in ALL, including financial Blood Adv., 2 (2018), pp. 3393-3403
National Institute for Health and Care Excellence Axicabtagene Ciloleucel for Treating Diffuse Large B-Cell Lymphoma and Primary Mediastinal Large B-Cell Lymphoma after 2 or More Systemic Therapies NICE (2019)
S. Johnson, et al. Cost-effectiveness of voretigene neparvovec-rzyl vs standard care for RPE65-mediated inherited retinal disease JAMA Ophthalmol., 137 (10) (2019), pp. 1115-1123
S. Viswanathan, A. Keating Overcoming the challenges of conducting translational research in cell therapy Front Med., 5 (2011), pp. 333-335
M. Papadaki Adaptation through collaboration: developing novel platforms to advance the delivery of advanced therapies to patients Front Med., 4 (2017), p. 56
S. Pearson Early Experience with Health Technology Assessment of Gene Therapies in the United States Pricing and Paying for Cures, Office of Health Economics (2019)
A. Kent, J. Spink Will rising prices and budget constraints prevent patients from accessing novel gene therapies? Gene Ther., 24 (2017), pp. 542-543
A. Bansal, et al. Estimating long-term survival for patients with relapsed or refractory large B-cell lymphoma treated with chimeric antigen receptor therapy: a comparison of standard and mixture cure models Med. Decis. Making, 39 (2019), pp. 294-298
M. Buessing, et al. Important considerations in modeling the cost-effectiveness for the first Food and Drug Administration-approved gene therapy and implications for future one-time therapies Value Health., 22 (2019), pp. 970-971
Jonsson, et al. Advanced therapy medicinal products and health technology assessment principles and practices for value-based and sustainable healthcare Eur. J. Health Econ., 20 (2019), pp. 427-438
T. Cowling, S. Jones Gene Therapy: International Regulatory and Health Technology Assessment (HTA) Activities and Reimbursement Status Canadian Agency for Drugs and Technologies in Health (2018)
L.P. Garrison, et al. Value-based pricing for emerging gene therapies: the economic case for a higher cost-effectiveness threshold J. Manag. Care Spec. Pharm., 25 (2019), pp. 793-799
S.P. Gavan, et al. Assessing the joint value of genomic-based diagnostic tests and gene therapies
J. Pers. Med., 9 (2) (2019), p. 28
D. Melchiorri, et al. Regulatory evaluation of Glybera in Europe – two committees, one mission Nat. Rev. Drug Discov., 12 (2013), p. 719
C. Remuzat, et al. Market access pathways for cell therapies in France J. Mark. Access Health Policy., 3 (2015)
C.L. Halioua-Haubold, et al. Regulatory considerations for gene therapy products in the US, EU, and Japan Yale J. Biol. Med., 90 (2017), pp. 683-693
J. Chisholm, et al. Current state of Health Canada regulation for cellular and gene therapy products: potential cures on the horizon Cytotherapy, 21 (2019), pp. 686-698
T. Bubela, et al. Recommendations for regulating the environmental risk of shedding for gene therapy and oncolytic viruses in Canada Front Med. (Lausanne)., 6 (2019), p. 58
M. Abou-El-Enein, et al. Overcoming challenges facing advanced therapies in the EU market
Cell Stem Cell., 19 (2016), pp. 293-297
EMA Committee for Advanced Therapies (CAT) Work Programme 2010 – 2015 EMA (2010)
N. Moran First gene therapy nears landmark European market authorization Nat. Biotechnol., 30 (2012), pp. 807-809
D. Cutler, et al. Insurance switching and mismatch between the costs and benefits of new technologies Am. J. Manag. Care., 23 (2017), pp. 750-757
J. Schimmer, S. Breazzano Investor outlook: solving gene therapy pricing…with a cures voucher? Hum. Gene Ther. Clin. Dev., 27 (2016), pp. 132-136
M. Elsallab, et al. Mitigating deficiencies in evidence during regulatory assessments of advanced therapies: a comparative study with other biologicals Mol. Ther. – Methods Clin. Dev., 18 (2020), pp. 269-279
K. Logviss, et al. Characteristics of clinical trials in rare vs. common diseases: a register-based Latvian study PLoS One, 13 (2018), Article e0194494
B.M. Buckley Clinical trials of orphan medicines Lancet, 371 (2008), pp. 2051-2055
S. Day, et al. Recommendations for the design of small population clinical trials Orphanet. J. Rare Dis., 13 (2018), p. 195
U.S. Department of Health and Human Services Food and Drug Administration Rare Diseases: Common Issues in Drug Development – Guidance for Industry FDA (2019)
H.P. Lesch, et al. Process development of adenoviral vector production in fixed bed bioreactor: from bench to commercial scale Hum. Gene Ther., 26 (2015), pp. 560-571
R.P. Harrison, et al. Chimeric antigen receptor-T cell therapy manufacturing: modelling the effect of offshore production on aggregate cost of goods Cytotherapy, 21 (2019), pp. 224-233
E. Fritsche, et al. Post-marketing safety and efficacy surveillance of cell and gene therapies in the EU: a critical review Cell Gene Ther. Insights., 5 (2019), pp. 1505-1521
J. Jorgensen, P. Kefalas Annuity payments can increase patient access to innovative cell and gene therapies under England’s net budget impact test J. Mark Access Health Policy, 5 (2017), Article 1355203
G.F. Pierce, D. Coffin The 1st WFH Gene Therapy Round Table: Understanding the landscape and challenges of gene therapy for haemophilia around the world Haemophilia, 25 (2019), pp. 189-194
P. Kefalas, et al. Establishing the cost of implementing a performance-based, managed entry agreement for a hypothetical CAR T-cell therapy J. Mark Access Health Policy., 6 (2018), Article 1511679
A. Boeke, et al. Vector production in an academic environment: a tool to assess production costs
Hum. Gene Ther. Methods., 24 (2013), pp. 49-57
D.N. Lakdawalla, et al. Defining elements of value in health care-a health economics approach: an ISPOR Special Task Force Report [3] Value Health, 21 (2018), pp. 131-139
M. Gutknecht, et al. A systematic review on methods used to evaluate patient preferences in psoriasis treatments J. Eur. Acad. Dermatol. Venereol., 30 (2016), pp. 1454-1464
J.K. Lin, et al. Cost effectiveness of chimeric antigen receptor T-cell therapy in relapsed or refractory pediatric B-cell acute lymphoblastic leukemia J. Clin. Oncol., 36 (32) (2018), pp. 3192-3202
Newdigs Focus Paying for Cures Toolkit Paying for Cures (2020)
M. Tempero Serving ‘a la CAR-T’: value-based pricing and gene therapy J. Natl. Compr. Canc. Netw., 15 (2017), p. 1179
R.M. Kirkner Must sky-high prices ‘come on down’ before the price is right? Manag. Care., 27 (2018), pp. 16-19
J.S. Sherkow CRISPR, patents, and the public health Yale J. Biol. Med., 90 (2017), pp. 667-672
M. White, et al. A guide to approaching regulatory considerations for lentiviral-mediated gene therapies Hum. Gene Ther. Methods, 28 (2017), pp. 163-176
K. Thompson, E.P. Foster The Cell Therapy Catapult: growing a U.K. cell therapy industry generating health and wealth Stem. Cells Dev., 22 (Suppl. 1) (2013), pp. 35-39
M. Schuessler-Lenz, et al. Regulators’ advice can make a difference: European Medicines Agency approval of zynteglo for beta thalassemia Clin. Pharmacol. Ther., 107 (2020), pp. 492-494
J. Tarnowski, et al. Delivering advanced therapies: the big pharma approach Gene Ther., 24 (2017), pp. 593-598
G. Bauer, et al. The path to successful commercialization of cell and gene therapies: empowering patient advocates Cytotherapy, 19 (2017), pp. 293-298
B. Klug, et al. Regulatory structures for gene therapy medicinal products in the European Union Methods Enzymol., 507 (2012), pp. 337-354
F. Bignami, et al. Participation of patients in the development of advanced therapy medicinal products Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz, 54 (2011), pp. 839-842
Alliance for Regenerative Medicine Regenerative Medicine & Rare Disease Alliance for Regenerative Medicine (2019)
J. Jørgensen, et al. Outcomes-based reimbursement for gene therapies in practice: the experience of recently launched CAR-T cell therapies in major European countries J. Mark Access Health Policy, 8 (2020), Article 1715536
A. Basu Financing cures in the United States Expert Rev. Pharmacoecon. Outcomes Res., 15 (2015), pp. 1-4
A. Hubert ATMPs in Europe: State of Play Alliance for Regenerative Medicine (2020)
US Food, Drug Administration Approved Cellular and Gene Therapy Products FDA (2019)
Canadian Agency for Drugs and Technologies in Health Axicabtagene Ciloleucel for Large B-Cell Lymphoma: Recommendations Canadian Agency for Drugs and Technologies in Health (2019)
S. Kirkey ‘What Price Life?’ At a Cost of Hundreds of Thousands a Dose, Novel Cancer-Killing Drug Sparks Debate National Post (2019)
Bluebirdbio Bluebird bio Announces Launch in Germany of ZYNTEGLO™ Bluebirdbio (2020)
R. Staines Germany First in EU to get Novartis’ SMA Gene Therapy, Costing almost 2m Euros Pharmaphorum (2020)
P. Hourd, et al. Manufacturing models permitting roll out/scale out of clinically led autologous cell therapies: regulatory and scientific challenges for comparability Cytotherapy, 16 (2014), pp. 1033-1047
P. Hourd, et al. Regulatory Challenges for the Manufacture and Scale-Out of Autologous Cell Therapies StemBook (2008)
J. Mansnérus Encountering challenges with the EU regulation on advance therapy medical products Eur. J. Health Law, 22 (2015), pp. 426-461
N. Touchot, M. Flume Early insights from commercialization of gene therapies in Europe Genes (2017)
Marsden, G. et al. (2017) Gene Therapy: Understanding the Science, Assessing the Evidence, and Paying for Value, ICER.
J.K. Lin, et al. Cost effectiveness of chimeric antigen receptor T-cell therapy in multiply relapsed or refractory adult large B-cell lymphoma J. Clin. Oncol., 37 (2019), pp. 2105-2119
R.R. Sarkar, et al. Cost-effectiveness of chimeric antigen receptor T-cell therapy in pediatric relapsed/refractory B-cell acute lymphoblastic leukemia J. Natl. Cancer Inst., 111 (7) (2018), pp. 719-726
M.D. Whittington, et al. Long-term survival and cost-effectiveness associated with axicabtagene ciloleucel vs chemotherapy for treatment of B-cell lymphoma JAMA Netw. Open, 2 (2019), Article e190035
J. Jorgensen, P. Kefalas Upgrading the SACT dataset and EBMT registry to enable outcomes-based reimbursement in oncology in England: a gap analysis and top-level cost estimate J. Mark Access Health Policy, 7 (2019), Article 1635842
C.R. Flowers, S.D. Ramsey What can cost-effectiveness analysis tell us about chimeric antigen receptor T-cell therapy for relapsed acute lymphoblastic leukemia? J. Clin. Oncol. (2018)
J.A. Roth, et al. Cost-effectiveness of axicabtagene ciloleucel for adult patients with relapsed or refractory large B-cell lymphoma in the United States J. Med. Econ., 21 (2018), pp. 1238-1245
R. Salzman, et al. Addressing the value of gene therapy and enhancing patient access to transformative treatments Mol. Ther., 26 (2018) 2717-2126
M. Zimmermann, et al. Cost utility of voretigene neparvovec for biallelic RPE65-mediated inherited retinal disease Value Health, 22 (2019), pp. 161-167
M.D. Whittington, et al. Long-term survival and value of chimeric antigen receptor T-cell therapy for pediatric patients with relapsed or refractory leukemia JAMA Pediatr., 172 (2018), pp. 1161-1168
E. Hanna, et al. Gene therapies development: slow progress and promising prospect J. Mark Access Health Policy., 5 (2017), Article 1265293
J. Jorgensen, P. Kefalas Reimbursement of licensed cell and gene therapies across the major European healthcare markets J. Mark Access Health Policy., 3 (2015)
N. Machin, et al. Gene therapy in hemophilia A: a cost-effectiveness analysis Blood Adv., 2 (2018), pp. 1792-1798
C. Morrison $1-million price tag set for Glybera gene therapy Nat. Biotechnol., 33 (2015), pp. 217-218
R. Kapoor, et al. Challenges in the gene therapy commercial ecosystem Nat. Biotechnol., 35 (2017), pp. 813-815
N.N. Malik Pay-for-performance pricing for a breakthrough heart drug: learnings for cell and gene therapies Regen. Med., 11 (2016), pp. 225-227
E. Flory, J. Reinhardt European regulatory tools for advanced therapy medicinal products Transfus. Med. Hemother., 40 (2013), pp. 409-412
Glossary
Allogeneic cells
cells derived from another person’s body (donor).
Autologous cells
cells derived from the patient’s own body.
Chimeric antigen receptor T cells (CAR-T)
genetically engineered T cells that can produce an artificial T cell receptor that can be used in onco-immunotherapy.
Discounting
mathematical procedure that calculates the current value of future costs and the health outcomes related to a product.
Gene therapy medicinal product (GTMPs)
a biological medicinal product that has the following characteristics: (a) contains an active substance that contains or comprises a recombinant nucleic acid used in or administered to humans with a view to regulating, repairing, replacing, adding, or deleting a genetic sequence; (b) its therapeutic, prophylactic or diagnostic effect relates directly to the recombinant nucleic acid sequence it contains, or to the product of genetic expression of this sequence. Gene therapy medicinal products do not include vaccines against infectious diseases (Directive 2001/83/EC).
Good Clinical Practices (GCP)
international ethical and scientific quality standard for the conduct of clinical trials in humans.
Good Manufacturing Practices (GMP)
international quality standard for the manufacturing of products in a consistent and controlled manner.
Health technology assessment (HTA)
process that systematically compiles the medical, social, and economic evidence as well as ethical issues related to the use of a health technology.