This paper was originally published by Ingrid Freije, Stéphane Lamouche, and Mario Tanguay in Therapeutic Innovation & Regulatory Science under a Creative Commons Attribution-NonCommercial 4.0 License (http://www.creativecommons.org/licenses/by-nc/4.0/)
Abstract
A 505(b)(2) application is a type of US new drug application (NDA) that contains full reports of investigations of safety and effectiveness, but where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. Most 505(b)(2) applications consist of changes to a previously approved drug product (ie, a new dosage form, new routes of administration, etc). Sponsors often face challenges determining the studies to be conducted to support approval via 505(b)(2) pathway. This 5-year (2012-2016) retrospective analysis reviewed approved 505(b)(2) NDAs available on the FDA website, to determine the nature of studies (preclinical, clinical pharmacology, and efficacy/safety) conducted for various types of submissions and to better understand the trends in terms of regulatory requirements. The database consisted of 226 NDAs. One hundred twelve of those 226 had complete FDA review information, with the following FDA submission classes being more prevalent: type 3, new dosage form; type 4, new combination; and type 5, new formulation or new manufacturer. Therefore, only these 112 NDAs were further examined as they could show trends in terms of the studies conducted for various types of applications. Based on the investigation of NDA review documents, coupled with guidance documents, decision trees for studies to be conducted have been developed, which may serve as a guide of recommendations for a successful 505(b)(2) development program and NDA submission.
Background
In 1999, the FDA published the Draft Guidance for Industry Applications Covered by Section 505(b)(2) which introduced this section of the Federal Food, Drug, and Cosmetic Act and described certain regulatory aspects such as eligibility, submission, and patent/exclusivity protection. By definition, the 505(b)(2) application is “an application that contains full reports of investigations of safety and effectiveness but where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference.”1 The 505(b)(2) is a pathway for new drug products that includes changes compared to an existing product, such as new formulations, routes of administration, and intended use. A drug submitted via 505(b)(2) can be approved based on data from studies not conducted by the sponsor, by relying on (1) Agency’s previous findings of safety and effectiveness (AFSE) of an approved drug; and/or (2) clinical and preclinical studies’ data from published literature without the right of reference. This requires not only a successful bridging to an RLD (reference listed drug), by the means of relative bioavailability (BA) or bioequivalence (BE) studies, but also some potential additional studies that may be needed to fully support efficacy and/or safety of the new product. If the sponsor has access to the originator data (ie, right of reference), then a supplemental NDA or the 505(b)(1) pathway would be applicable for the previous changes.
In light of the recent growing interest in the 505(b)(2) pathway, the industry finds itself looking for guidance on the specifics of the clinical development program of these NDAs. In addition to the specific FDA draft guidance on 505(b)(2), the FDA published a draft guidance on “Determining Whether to Submit an ANDA or a 505(b)(2) Application” that guides sponsors on the type of submissions (generic vs 505(b)(2)) that should be applicable to their specific drug, and an article that provides an overview of some clinical pharmacology aspects seen in approved 505(b)(2) NDAs.2,3 However, these guides do not provide details regarding drug development aspects. Since 505(b)(2) applications are considered NDAs, review of some US requirements described in the 21 CFR (Code of Federal Regulations) Title 21, Ch. I (ie, Part Sections 320.27; 320.24; 320.25; 320.22; 300.50; 314.50)4 and various FDA guidance documents provides valuable information regarding the design and conduct of studies submitted in NDAs in general.5–13 It is critical to understand FDA’s expectations in order to facilitate discussion with the Agency through pre-IND or other FDA meetings, and to avoid deficiencies in terms of data supporting approval for a new product submitted under the 505(b)(2) pathway.
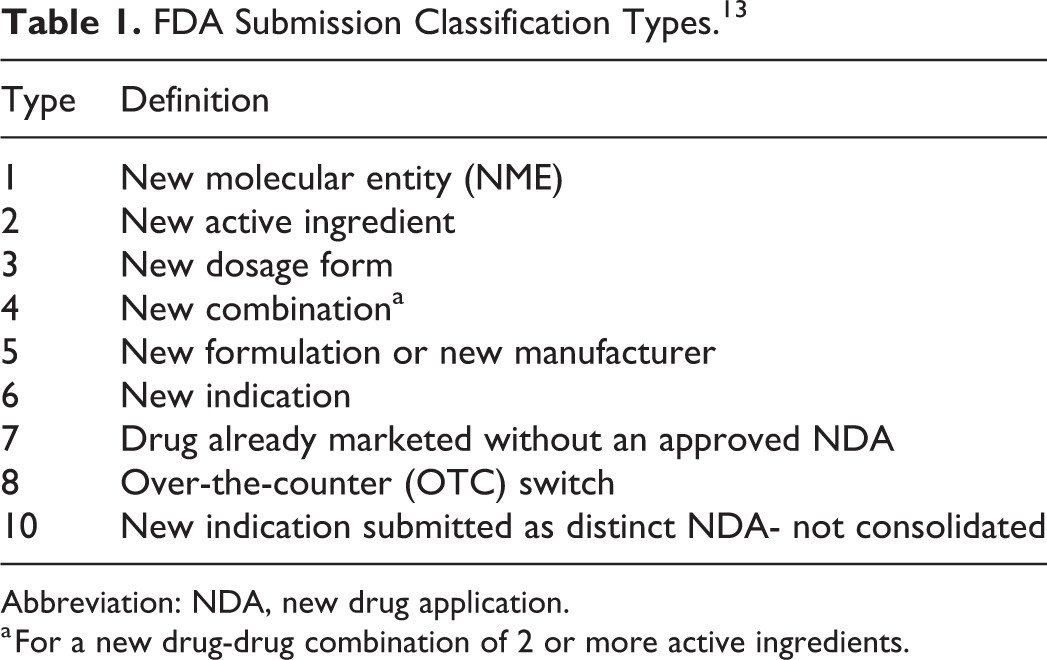
This retrospective analysis aims to provide not only an overview of recently approved 505(b)(2) NDAs (2012-2016), but also to summarize the trends in the studies conducted to support NDA approval. It is important to recognize that, without a thorough understanding of the regulatory history, the clinical studies for each product may have been done to meet (1) FDA requirements, (2) other agency requirements, and (3) sponsor choice. However, upon the particular type of modifications made in the new drug product as compared to an RLD, as defined by the FDA submission classification type (Table 1),13 the recommendations concerning the preclinical, clinical, and especially the clinical pharmacology studies to support a 505(b)(2) NDA will be discussed. The ultimate objective of this review is to allow an easier navigation through the various types of 505(b)(2) submissions for drug development organizations.
Table 1 FDA Submission Classification Types
Materials and Methods
For the first part of this retrospective analysis, a search of the FDA website was conducted to retrieve all 505(b)(2) NDAs approved between January 2012 and December 2016, from “NDA and BLA Calendar Year Approvals.”14 In addition, information regarding brand names, FDA submission classification type, dosage form, and routes of administration, were collected from FDA’s Approved Drug Products database (drugs@FDA) to provide a general overview of 505(b)(2) approvals across the last 5 years, complemented by information from the FDA’s “Orange Book” database, if needed.15,16
For the second part, an in-depth review of the selected products’ Drug Approval Package was performed, with an emphasis on the following NDA review sections: Summary, Medical, Pharmacology, and Clinical Pharmacology Biopharmaceutics. Only products with available reviews were included. A database was built including information about the RLD, the type of clinical pharmacology studies, the presence or absence of preclinical and clinical/efficacy or safety studies, as well as the Agency’s recommendations for approval communicated during pre-IND or pre-NDA meetings. Only NDAs for submission classifications prevalent enough to identify trends in the types of studies were retained in this part of our investigation and in the final discussion. This review article will highlight the nature and rationale behind the studies conducted, and provide decision trees that were created based on the trends observed for various types of oral drug products approved via the 505(b)(2) regulatory pathway, and further review of various FDA guidance documents.
Results and Discussion
General Overview of 505(b)(2) NDAs Approved From 2012 to 2016
A total of 226 505(b)(2) NDAs were approved over this period of 5 years, and the number of approvals was generally consistent year to year (Figure 1).
Figure 1. Number of 505(b)(2) approvals per year from 2012 to 2016.
Of the 226 NDAs, FDA provided the submission classification types in 224 cases, which are illustrated in Figure 2. The following FDA submission classes were the most prevalent: type 5 (new formulation or new manufacturer; 43.3%); type 3 (new dosage form; 28.6%); followed by type 4 (new combination; 12.9%). New molecular entities and new active ingredients (type 1 and 2, respectively) were rare, most likely because such products would typically be submitted as 505(b)(1) NDAs. This classification tool allowed the researchers to filter and better understand the nature of the drug products submitted via the 505(b)(2) pathway. However, even though a specific category was assigned by the FDA, in some cases, various modifications could be seen (eg, type 3 products could include a changed formulation, a new route of administration, an altered excipient, or a new strength).
Figure 2. FDA submission classification of drug products approved via 505(b)(2) pathway from 2012 to 2016 (n = 224). Refer to Table 1 for FDA submission classification types.
Studies Submitted for the Most Prevalent Types of 505(b)(2) Products
Out of the 226 NDAs previously described, 181 had Drug Approval Packages available on the FDA website. In addition to that, out of these entries, 112 NDAs were of a submission classification type prevalent enough to show trends with regard to the type of studies submitted by the sponsors, and were retained for this part of the discussion (ie, types 3, 4, and 5). In fact, 51 NDAs were excluded because of their low prevalence with respect to type (Figure 2) and nontypical case-by-case 505(b)(2) NDAs (n = 5). Moreover, 18 NDAs submitted as type 7 were not included in this review as these did not represent a change to an RLD.
Table 2 identifies the types of studies conducted for the selected 505(b)(2) applications relative to the submission classification type for types 3, 5, and 4 (new dosage form, new formulation, or new combination respectively).
Table 2. Prevalence of Various Studies Conducted for Different Types of 505(b)(2) NDAs.a
The following studies were only found very rarely in the reviewed NDAs: pharmacokinetic (PK) studies in patients with hepatic impairment or renal impairment; QT prolongation studies; drug-drug interaction (DDI) studies evaluating interactions of the drug product with either enzymatic or transporter inhibitors/inducers, or with commonly used concomitant drug; preclinical toxicology, genotoxicity, carcinogenicity, and reproductive toxicity studies. These studies were not included in Table 2 as they were requested by the Agency to address preexisting deficiencies in the label of the RLD. Therefore, the following discussion will rather focus on the additional studies frequently requested to support modifications to RLDs.
Choice of the RLD
A scientific bridge to a relevant RLD is required if the sponsor relies on the AFSE of an already approved drug. An RLD is chosen based on available drugs that contain the same active pharmaceutical ingredient (API). A side-to-side comparison of labels identifies the most appropriate RLD (eg, in terms of route of administration or formulation), while keeping in mind that the level of similarity between the proposed product and the RLD could have an impact on the number of studies to be conducted. However, the Agency should agree with the choice of the RLD before beginning the process of the new drug development. In some cases, when the selected RLD was no longer available (eg, discontinued for reasons other than safety and effectiveness), the comparator drug was a product approved via the ANDA pathway, and which was designated as the reference standard in the Orange Book. However, safety and efficacy data were derived from the original NDA of the discontinued product in such cases. In certain situations, it might be necessary to use multiple RLDs during the development of the proposed product (eg, for fixed-dose combination [FDC] products where sponsors are combining two already approved oral drug products). It may also be required to conduct bridging studies with different RLDs in order to seek multiple indications. As an example, methotrexate auto-injector for subcutaneous administration (NDA 205776) required two bridging studies: one for BA/BE compared to methotrexate intramuscular injection RLD, and one to obtain approval for additional indications specific to the methotrexate sodium oral tablet RLD.15
Bridging approaches
In 69.6% of these NDAs (n = 112), a scientific bridge was established by a single-dose BA/BE study comparing PK of the new product and RLD (Table 3), which is in line with the FDA Draft Guidance for Industry on Applications Covered By Section 505(b)(2).1 Even though a relative BA study was performed, BE did not necessarily have to be demonstrated. In fact, there are situations where the proposed product is expected to have a rate and/or extent of BA (ie, maximal concentration [Cmax] and/or area under the curve [AUC]) different than the RLD, because of the nature of the formulation change (eg, new extended-release [ER] product), or because an improvement in terms of PK characteristics is anticipated (eg, drug with a more rapid onset of action). On the other hand, any differences in BA had to be typically supported by additional clinical trials. If the exposure was lower than the RLD, additional studies could be required to confirm efficacy of the new product. In contrast, additional studies could be required to support safety if the exposure was higher than the RLD. For example, a new meloxicam capsule with reduced particle size (NDA 207233) was developed in order to obtain a more rapid absorption, which was demonstrated by time to peak concentration that was 2 hours earlier than the RLD (meloxicam tablet).15 However, as this resulted in a 33% reduction in Cmax, the sponsor had to conduct a clinical trial to confirm that efficacy and safety of the new product in the targeted population was still equivalent to that established by the RLD.
Table 3. Bridging Approaches.
As shown in Table 3, other scientific bridging approaches were also occasionally adopted. For example, a PK/PD study may serve as a bridging approach to support the approval of a new drug delivery system (eg, a new modified-release [MR] dosage form of an immediate-release [IR] RLD) that may show some differences in PK, as compared with the RLD.5 A good understanding of the exposure-response relationship is then required to demonstrate that the difference in BA would not lead to a different therapeutic outcome. When a PD endpoint can be considered as surrogate marker of the clinical outcome, such a PK/PD approach may waive the need for additional efficacy/safety trials. For example, for a new cysteamine bitartrate delayed-release (DR) capsule (NDA 203389), the effect on reduction in white blood cells was compared to the IR RLD in healthy volunteers.15 In certain situations, sponsors can also demonstrate the concentration-response relationship in vitro, in lieu of an in vivo assessment. That was the case of argatroban IV injection (NDA 206769), where equivalence with the RLD was established in vitro by measuring the anticoagulant PD activity (ie, prothrombin time) in pooled donor human plasma spiked with clinically relevant concentrations of both products individually.15
Nevertheless, in 20% of NDAs, sponsors have referenced an RLD and relied on the AFSE without conducting any bridging studies to the RLD in question. Table 4 shows the rationales that can support an in vivo BA/BE waiver (biowaiver).
Table 4. Rationales for in Vivo BA/BE Waiver.
In vivo BA/BE waivers were rather requested on either two major bases. First, as per the FDA Draft Guidance for Industry “Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System,” a demonstration of in vivo BA/BE may not be necessary for drug products containing class 1 (high solubility–high permeability) and class 3 (high solubility–low permeability) drug substances, as long as the inactive ingredients used in the dosage form do not significantly affect absorption of the API.6
Second, biowaivers may also be justified as per 21 CFR 320.22(b) entitled “Criteria for Waiver of Evidence of In Vivo Bioavailability or Bioequivalence,” where the BA/BE of a drug product may be self-evident.4 Among the NDAs that were reviewed, these criteria were applied for parenteral solution (n = 19) and for oral solutions (n = 3). However, in these cases, the Agency recommended that sponsors provide adequate justifications with regard to any differences between the proposed product and the RLD (eg, components and their composition, indication, stability, instructions for dilution, pH and osmolality, delivered volume, and infusion rate), and their potential impact on clinical safety and efficacy, such as in NDA 207131 for cefazolin sodium intravenous product.15
Food-effect studies
For almost all 505(b)(2) NDAs of orally administered products, regardless of the changes made (Table 2), a study evaluating the effect of food on the BA of the new drug was conducted. In some rare cases of IR and MR NDAs, BE studies (Test vs RLD) under both fasting and fed conditions have been conducted, as typically recommended for products submitted as ANDAs.9 However, as per the “Guidance for Industry on Food-Effect Bioavailability and Fed Bioequivalence Studies,” for NDAs, including 505(b)(2)s, a BA/BE study under fasting conditions (Test vs RLD) and a food-effect study on the new product (fast vs fed) would usually be recommended.9
Based on the experience of our own organization, it is also possible to somehow streamline the development by combining the fasting BA/BE assessment with the food-effect evaluation, through a 3-way crossover trial, that is, test product and the RLD administered in fasting conditions, with an additional third arm for the test product administered under fed conditions, thereby providing the fast and fed comparison. The Agency typically accepted this approach, which may save time and reduce costs of development.
Clinical efficacy/safety studies
In the case of 50 of 112 NDAs, sponsors could not rely solely on the AFSE of the RLD and conducted a clinical efficacy/safety study in addition to the BA/BE study.
The Agency stated in many reviews such as in NDA 204768, indomethacin oral capsule, that “when BE criteria is not met, the number of adequate and well controlled phase 3 efficacy studies will depend on the similarities in the concentration-time curves of the reformulated drug compared to RLD.”15 Otherwise, proper justification for the lack of such studies could be provided, such as a large therapeutic index or a good understanding of the PK/PD relationship.
However, even when BA/BE criteria are met, a clinical efficacy/safety study is still usually required in the situations presented in Table 5.
Table 5. Reasons for Clinical Efficacy/Safety Studies.
In the case of new FDC products, a demonstration of the therapeutic advantage of the FDC over each monotherapy should be demonstrated, as required by the combination rule stated in 21CFR 300.50(a).4 This information can be acquired from published literature or from previous NDAs. For example, in the case of the oral perindopril arginine and amlodipine FDC product, NDA 205003, which was already approved in Europe, the FDA considered that the referenced studies did not support superiority of the combination to the highest doses of each component of the monotherapies.15 As a result, a phase 3 clinical trial comparing the highest proposed dose of the combination with the highest approved doses of perindopril erbumine and amlodipine, administered as monotherapies, was conducted to show statistical and clinical superiority. In addition, when the dosing regimen of the FDC product differs from that of each single RLD (eg, once daily vs twice daily), a non-inferiority posology bridging has been required to fulfill the regulatory requirement, and to justify the appropriateness of reliance on data from the RLDs. As an example, in the case of a new canagliflozin/metformin FDC product (NDA 204353), the sponsor was asked to demonstrate that the efficacy of canagliflozin would not be altered when administered once-daily as compared to the previously recommended twice-daily regimen, by either using a robust modeling and simulation strategy, or a clinical efficacy trial comparing both dosing regimens.15 In that specific case, the sponsor chose to conduct a clinical trial.
Other types of safety studies were also occasionally observed, such as evaluation of abuse-deterrent properties (ie, human abuse liability studies); additional dermal safety studies for new topical products (ie, evaluation of sensitization, photosensitization/photoallergy, cumulative irritancy, and static sun protection factor [SPF] value of the vehicle); and safety assessment of the oropharyngeal area for new orally disintegrated tablets (NDA 208025, 204326).15
Preclinical studies
In some cases, sponsors conducted additional preclinical studies (ie, safety, carcinogenicity/genotoxicity, reproductive and/or drug-drug combination evaluations) for new formulations or new routes of administration (n = 11), for products with altered excipients (n = 8) and for new oral FDC products (n = 6). Table 6 summarizes the additional preclinical studies that were conducted to support the lack of adequate published literature or prior human data of intended changes in regard to API(s), excipients, route of administration, and proposed combination.
Table 6. Situations Where APIs, Excipients, or Route of Administration Considerations May Require Additional Nonclinical Studies.
For example, in 6 NDAs of products administered via a new route of administration (3 parenteral, 1 ophthalmic, 1 topical, and 1 transdermal) that had oral RLDs, sponsors conducted a preclinical pharmacology/toxicology bridging study to support safety of the new clinical route.
Moreover, in the case of new formulations, sponsors evaluated the adequacy of the current safety database to support not only the added excipients in their new formulations, but also the effect of those excipients on the clinical exposure of the API. In the case of NDA 208090 (a new ER oxycodone capsule), additional preclinical studies (ADME studies, chronic toxicity, reproductive toxicity, and carcinogenicity studies) were required to address the lack of PK data and to assess the clinical exposure of the API with the new excipients.15
Finally, for oral FDC products, safety data for each mono-product was sufficient in general to support the following aspects of the combination: proposed indication, dose, duration of treatment (ie, chronic or acute use), and targeted population. However, the lack of prior human safety experience with the new proposed combination had to be evaluated, especially when both APIs could elicit PD activities and toxicity on similar vital systems. For example, for NDA 206439 (memantine hydrochloride ER + donepezil hydrochloride), the sponsor conducted additional preclinical studies in order to evaluate toxicity, dose-ranging, and suspected synergistic effect of both products on the central nervous system.15
Multiple-dose BA/BE and alcohol-dose dumping in vitro
A multiple-dose PK study allows assessing the rate and extent of absorption of the API into the systemic circulation when the levels of the drug substance have reached steady state. In general, a single-dose PK study is considered as a more sensitive method to determine the BA/BE of a drug. Therefore, most of the BA/BE bridging studies included in 505(b)(2) NDAs were done in a single-dose fashion. Nevertheless, 20 NDAs included a multiple-dose BA/BE in addition to the single-dose BA/BE study. These involved new MR formulations, which include ER and DR drug products.17,18 The draft FDA Guidance on “BA and BE Studies Submitted in NDAs or INDs—General Considerations” describes when a multiple-dose BA/BE study might be required and includes the case of ER dosage form.18 In fact, attainment of steady state is often needed in order to robustly compare the relative PK exposure (eg, AUC over 24 hours) of the new ER formulation versus the IR or another MR RLD, especially when the dosing regimens are different.
As for DR products, the same draft FDA guidance states that, in general, demonstration of BA/BE for such products is the same as for an IR product, meaning a single-dose BA/BE under fasted conditions along with a food-effect study.18 Nevertheless, in the case of the 5 DR NDAs that were included in our review, sponsors also conducted a multiple-dose BA/BE study. The rationale behind this is not clear, but it could be attributed to the complex release characteristics of the DR products.18
Moreover, in order to validate the ER claim, sponsors should conduct an in vitro study to rule out the possible occurrence of alcohol-induced dose dumping (ie, unintended and rapid drug release in a short period of time of the entire amount of the drug contained in the ER dosage form), which could jeopardize the safety of patients.19 The Agency may also request an in vivo BE study with concomitant administration of alcohol, if in vitro results show a positive dose dumping.20
Dose-proportionality studies
Dose-proportionality studies evaluate if increases in the administered dose are accompanied by proportional increases in exposure (AUC or Cmax). The FDA Guidance for Industry on “Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs—General Considerations” clarifies that for ER dosage forms containing drugs with nonlinear PK over the therapeutic dose range, a single dose of the highest and lowest strengths of the ER product should be compared to their corresponding IR references administered over the ER dosing interval.18 In addition, when the new drug product strengths are not proportionally similar in composition (ie, different ratios of active/inactive ingredients), a single-dose fasting dosage strength equivalence assessment study or a dosage strength proportionality study should be conducted.18 Dose-proportionality studies were conducted in the case of 15 NDAs where sponsors requested additional approval for multiple strengths for their new dosage form. Therefore, they either conducted a dose proportionality study, or a BA/BE study with each strength, compared to the respective RLD.
However, in other NDAs with additional strengths, sponsors requested a waiver for lower strengths on the basis of adequate in vitro dissolution tests, or if in vivo BA/BE was self-evident as per 21 CFR, section 320.22.4
Drug-drug interaction studies
In the case of 12 of 18 NDAs of FDC products, PK DDI studies were conducted to evaluate the possible interaction between the two combined drugs. Even though there is no specific FDA guidance on oral FDC products that include two already approved drugs, NDA reviews clearly indicated that DDI studies should be conducted between the therapeutic components of the FDC if this was not conducted previously, and if the potential for an interaction cannot be ruled out. Conduct of such a study is also a recommendation from the FDA Guidance for Industry on “Fixed Dose Combinations, Co-Packaged Drug Products, and Single Entity Versions of Previously Approved Antiretrovirals for the Treatment of HIV.”7
Trends in Studies for New Oral Formulations and New Oral FDCs
All regulatory and scientific requirements relative to preclinical, clinical pharmacology, and clinical efficacy/safety disciplines should be fulfilled in the NDA submission. This comprehensive review of NDAs approved over 2012-2016 has revealed trends in the types of studies that were conducted per the most frequent changes to an RLD (ie, new formulation and new oral FDC).
New oral formulation
In this case, sponsors applied for the 505(b)(2) pathway for a change in the drug product either from one IR dosage form (RLD) to another, or from one dosage form (RLD) to an ER dosage form. In terms of clinical pharmacology studies, 88% of NDAs included a single-dose BA/BE study and a food-effect study. However, for new ER formulations, a multiple-dose BA/BE study and an in vitro alcohol dose-dumping study (72%) were also performed. A dose proportionality study was also conducted in 18% of NDAs, and 34% of NDAs included a clinical efficacy/safety study.
New oral FDC
In this case, both RLDs could be of the same dosage form (IR/IR or MR/MR) or two different dosage forms (IR/MR).
Depending on the dosage form of the oral FDC (IR or MR), either the conditions for new IR formulations or those for new ER formulations listed above under “New Oral Formulation” should be considered for clinical pharmacology studies. However, in both cases, a crossover BA/BE study comparing the FDC to equal doses of the individual APIs should be conducted.
Overall, 17 out of 18 NDAs reviewed for oral FDCs included a clinical efficacy/safety study to support the proposed indication.
Decision Trees
Any applicant who wishes to go through the 505(b)(2) route should seek the Agency’s advice pre-emptively with regard to their development plan. Nevertheless, some general recommendations can still be made. Based on the trends that were observed during this extensive review of 505(b)(2) applications, and on the parallel examination of various applicable FDA guidelines, decision trees are proposed to help determine what type of clinical trials should be conducted for the most prevalent types of change to an approved RLD (Figures 3–6).
Figure 3. Clinical pharmacology decision tree for new immediate-release (IR) formulations. *With regards to the biowaiver, some sponsors may still prefer to conduct at least one BA/BE study, even if their product would be eligible for a biowaiver (e.g., for global marketing considerations).Figure 4. Clinical pharmacology decision tree for new extended-release (ER) formulations.Figure 4. Clinical pharmacology decision tree for new extended-release (ER) formulations.Figure 5. Clinical efficacy/safety decision tree for change in oral formulation. *The number of adequate and well controlled phase 3 efficacy studies will depend on the similarities in the concentration-time curves of the reformulated drug compared to RLD. A PK/PD study can possibly replace a clinical efficacy study if the PK/PD relationship of the study drug is well understood.5Figure 6. Clinical efficacy/safety decision tree for new oral fixed-dose combinations (FDCs). *The number of adequate and well controlled phase 3 efficacy studies will depend on the similarities in the concentration-time curves of the reformulated drug compared to RLD.
Conclusion
The 505(b)(2) pathway has become a mainstream in the pharmaceutical development industry. This comprehensive review of NDAs approved over 2012-2016 has revealed trends in studies conducted per the most frequent changes to an RLD. While it is true that there is no specific guidance on 505(b)(2) drug development programs, most FDA guidance documents referring to NDAs are relevant in understanding not only the rationale of the studies that were conducted but also in pointing out what needs to be done in order to get an approval via the 505(b)(2) pathway. The decision trees constructed following this review can inform development strategies to be presented and confirmed by FDA in meetings with the Agency.
Author Note
This paper has been presented at the American Association of Pharmaceutical Scientists (AAPS) Annual Meeting on the 13th of November, 2017, in San Diego, CA.
Acknowledgments
The authors would like to thank the following colleagues for their critical review: Susan Watts, Judith Ng-Cashin, and Osama Al-Jawadi.
Declaration of Conflicting Interests
No potential conflicts were declared.
Funding
No financial support of the research, authorship, and/or publication of this article was declared.
References
US Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER) . Draft guidance for industry applications covered by section 505(b)(2). https://www.fda.gov/downloads/Drugs/Guidances/ucm079345.pdf Published October 1999. Accessed June 1, 2017.
Sheetal, A, Wei, Q, Chadrahas, Q. Overview of recently approved 505(b)(2) new drug applications (2010-2012): role of clinical pharmacology. J Clin Pharmacol. 2014;54(12):1330–1336.
US FDA, CDER . Guidance for Industry. Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. https://www.fda.gov/downloads/Drugs/Guidances/ucm070246.pdf. Published December 2017. Accessed June 3, 2017.






















(2) Pathway: Uncovering Drug Development Trends and Regulatory Requirements")


(2) Drug Patent Approval Process: Uses and Potential Advantages")

(2) Regulatory Pathway for New Drug Applications: An Overview on the Advanced Formulation Approach and Challenges")





(2) Approval")

