This paper was originally published by Martin Schiestl, Markus Zabransky, and Fritz Sörgel in Drug Des Devel Ther. 2017; 11: 1509–1515 under a Creative Commons Attribution – Non Commercial (unported, v3.0) License.
Abstract
A biosimilar is defined by the European Medicines Agency as a biological medicine that is similar to another biological medicine that has already been authorized for use. A science-based regulatory framework to ensure high-quality biosimilars has been established in Europe since 2005 and is monitored and updated on an ongoing basis. The guiding principle of a biosimilar development program is to establish similarity between the biosimilar and the reference medicine by the best possible means, ensuring that the previously proven safety and efficacy of the reference medicinal product also applies to the biosimilar. Development of a biosimilar is underpinned by state-of-the-art analytical techniques to characterize both reference medicines and biosimilars. The extent and nature of the nonclinical in vivo studies and clinical studies to be performed depend on the level of evidence obtained in the previous step(s), including the robustness of the physicochemical, biological, and nonclinical in vitro data. Extrapolation is an important element of the biosimilarity concept. When biosimilar comparability has been demonstrated in one indication, extrapolation of the data package to other indications of the reference medicine could be acceptable, but needs to be scientifically justified and considered in light of the demonstrated level of sameness by all analytical, nonclinical, and clinical data. The credibility of the scientific basis behind the biosimilar concept, and quality of regulatory decision-making, is demonstrated by the successful approval and clinical use of 20 biosimilar medicines since 2006 when Omnitrope® was the first biosimilar to be approved. The regulatory environment for biosimilars continues to evolve, both in recognition of advances in technology/analytical methods and the availability of new targets for biosimilar development.
Unlike small-molecule drugs, which can be chemically synthesized, biologic drugs are produced by living cells and require advanced manufacturing and production processes.1 At the molecular level, they are typically large recombinant proteins that undergo complex post-translational modifications. Such complex molecules are typically expensive to develop and to produce. They also lack direct market competition before the advent of biosimilars. As a result, biologic agents have a high per-unit acquisition cost, which results in restricted access for many patients.2,3 The expiry of data protection/patents for the first original biotherapeutics led to the development of biosimilars, defined by the European Medicines Agency (EMA) as a biological medicine that is similar to another biological medicine that has already been authorized for use.4 A science-based regulatory framework to ensure high-quality biosimilars has been established in the European Union (EU) since 2005 and is monitored and updated on an ongoing basis.5 The pathway requires manufacturers to demonstrate similarity to a “reference” biologic (typically the originator) in terms of safety, efficacy, and quality, but not through a clinical research program of the scale that is demanded for initial approval of an originator biologic.6 This ultimately means biosimilars are less expensive to develop and acquire than their reference medicines, creating economic competition that has the potential to break monopolies, expand patient access to treatment, and release funds to support future innovation and research.7–9 Understandably, the development of the biosimilar regulatory pathway was opposed by various stakeholders with an interest in originator biologics. However, it is interesting to note that many of those who opposed biosimilars have now entered the biosimilars space, recognizing that biosimilars and innovator pharmaceuticals can co-exist.10,11
The first biosimilar medicine, Omnitrope® (biosimilar recombinant human growth hormone [rhGH]; Sandoz, Kundl, Austria), was approved in Europe by the EMA in 2006.12 Since then, 20 biosimilars have been approved in Europe; these medicines are based on hematopoietic growth factors (such as filgrastim and epoetin), insulin, follitropin, and monoclonal antibodies (such as infliximab and etanercept). This article reviews the emergence of the biosimilar regulatory framework and its evolution over the last decade.
Emergence of the biosimilar regulatory framework
Biosimilars draw on scientific principles that have been used for many years by the pharmaceutical industry and its regulators. Manufacturing processes for biologics are often changed for a variety of reasons, including scaling up of the process, improving efficiency, or modernization when equipment needs to be updated or replaced.13 To allow such manufacturing changes to occur without the need for companies to conduct a new clinical development program, regulators devised the comparability concept to establish whether the pre- and postchange products were sufficiently similar to permit ongoing marketing under the same product label.
The International Conference on Harmonization Q5E tripartite comparability guidelines, used in Europe, the USA, and Japan, state that the goal of the comparability exercise is to provide analytical evidence that a biological medicine has highly similar quality attributes (molecular structure) before and after manufacturing process changes.14 If differences are observed, they must be shown to have no adverse impact on safety, efficacy, or immunogenicity. If a manufacturer can provide assurance of comparability through analytical studies alone, nonclinical or clinical studies with the postchange product are not warranted. However, where the relationship between molecular attributes and safety and efficacy has not been established and analytical differences are observed, it might be appropriate to perform a combination of analytical, nonclinical, and/or clinical studies. These same principles are used to show that biosimilars have no clinically meaningful differences to their reference medicines, and thus form the basis of the European biosimilars regulatory pathway.
The evolving regulatory environment for biosimilars in the EU
The EU was the first region in the world to define a policy and legal framework for the approval of biosimilar medicines. The start point was possibly a 1998 concept paper on the development of a guideline on the comparability of biotechnology-derived products;15 subsequently, the idea of a “similar biological medicinal product” was first introduced into EU legislation in 2001 (Directive 2001/83/EC; a directive is a legal act of the EU).16 In 2003, Annex 1 was incorporated into the Directive to make the process for marketing authorization and preparation of biosimilar medicinal products clearer and more precise.17 Annex 1 stated that in addition to using pharmaceutical, chemical, and biological data to demonstrate biosimilarity, bioequivalence and bio-availability data should also be provided. In addition, the type and amount of additional data (ie, toxicological and other nonclinical and appropriate clinical data) would be determined on a case-by-case basis, taking into account the specific characteristic of each individual medicinal product. The Directive was further revised in 2004 with the inclusion of a clause that allowed the full development of biosimilars before the originator’s patent had expired.18
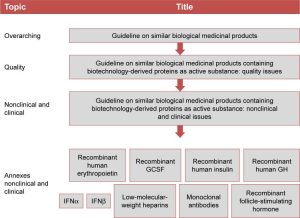
With the legal framework for biosimilars having been established, the EMA, together with the Committee for Medicinal Products for Human Use (CHMP), the Biotechnology Working Party, and the Working Party on Similar Biological Medicinal Products released specific guidelines to deal with all aspects of the development, production, testing, and regulation of biosimilar medicines. The EMA issued an overarching biosimilars guideline in 2005, which was followed in 2006 by two more specific guidelines on quality, clinical, and nonclinical issues relating to the development of biosimilars; these were recently updated.5,19,20 Product-specific guidelines and guidance on the assessment of immunogenicity are also available (Figure 1).
Figure 1: Overview of EMA guidelines related to the development and approval of biosimilars. Abbreviations: EMA, European Medicines Agency; GCSF, granulocyte colony-stimulating factor; GH, growth hormone; IFNα, interferon alpha; IFNβ, interferon beta.
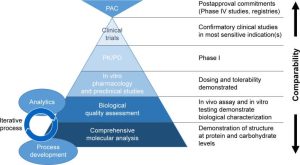
Essentially, the guiding principle of a biosimilar development program is to establish similarity between the biosimilar and the reference medicine by the best possible means, ensuring that the previously proven safety and efficacy of the reference medicinal product also applies to the biosimilar. Requirements for the development and approval of a biosimilar are different to those of a traditional, chemically derived generic version of a small-molecule drug; for the latter, typically only pharmacological bioequivalence data are required by regulatory bodies. Development of a biosimilar is underpinned by state-of-the-art analytical techniques to characterize both reference medicines and biosimilars (Figure 2). EMA requires sponsors to demonstrate high similarity between both medicines for all physicochemical and biological attributes.20 Developing a biosimilar manufacturing process can, therefore, involve multiple iterations in early-stage development and takes more time than is required for an originator medicine at the same stage of development.13 A stepwise approach is normally recommended throughout the development program, starting with comprehensive physicochemical and biological characterization, whereby different assays are often used to assess the same attribute in an orthogonal approach (Table 1). One trend of the last 10 years has seen nonclinical in vivo testing replaced where possible by in vitro assays to reflect changes in animal protection legislation.21 The extent and nature of the nonclinical in vivo studies and clinical studies to be performed depend on the level of evidence obtained in the previous step(s), including the robustness of the physicochemical, biological, and nonclinical in vitro data. Generally, the aim of clinical data is to address slight differences shown at previous steps and to confirm comparable clinical performance of the biosimilar and the reference medicine; therefore, studies should be sensitive enough with regard to design, conduct, end points, and/or population to detect such differences.19 Interestingly, continued advances in analytical techniques have led to the suggestion that a combination of analytical characterization, comparative pharmacokinetic, and postmarketing monitoring may provide more discriminatory evidence than large preapproval studies to demonstrate therapeutic equivalence.22
Figure 2: The biosimilar development process. Abbreviations: PAC, postapproval commitments; PD, pharmacodynamic; PK, pharmacokinetic.
Table 1
Examples of analytical methods for characterizing molecular attributes of a biologic
Molecular attribute
Methods
Amino acid sequence, primary structure
Peptide mapping (LC–MS)
Peptide mass fingerprint (MALDI-MS)
MALDI TOF
MS amino acid sequencing
Higher order structure, conformation
Far and near UV CD spectroscopy
Thermal stability
NMR
SPR
ELISA
Post-translational modifications
Mass spectrometry
NP-HPLC–(MS)
GC–MS
HPAEC-PAD
Polarity, charge isoforms
RP-HPLC
CZE
Size, detection of aggregates
SDS-PAGE
HP-SEC
AF4
AUC
Binding
Cell-based assays
SPR
ELISA
Biological activity
Cell assays
In vivo assay
Abbreviations: AF4, asymmetrical flow field-flow fractionation; AUC, analytical ultracentrifugation; CZE, capillary zone electrophoresis; ELISA, enzyme-linked immunosorbent assay; GC–MS, gas chromatography–mass spectroscopy; HPAEC-PAD, high performance anion-exchange chromatography with pulsed amperometric detection; LC–MS, liquid chromatography–mass spectroscopy; MALDI, matrix-assisted laser desorption/ionization; MS, mass spectroscopy; NMR, nuclear magnetic resonance; NP-HPLC (MS), normal phase high-performance liquid chromatography with optional mass spectrometry; RP-HPLC, reverse-phase high-performance liquid chromatography; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; SEC, size exclusion chromatography; SPR, surface plasmon resonance spectroscopy; TOF, time of flight; UV CD, ultraviolet circular dichroism.
The development of regulatory guidelines for biosimilars was largely informed by experience with biosimilar rhGH. In 2003, data from analytical, nonclinical, and clinical studies comparing liquid and lyophilized biosimilar rhGH formulations with its reference medicine, Genotropin® (Pfizer), were submitted to the CHMP for review.
Using state-of-the-art analytical procedures, the drug substance for biosimilar rhGH was thoroughly characterized with regard to its physicochemical properties and purity profile, and shown to be highly similar to the reference medicine in all methods applied. The results of a number of animal studies confirmed the pharmacodynamic (PD) properties of biosimilar rhGH, comparable potency between biosimilar rhGH and the reference medicine, and no relevant toxic effects. Local tolerance studies in rabbits further confirmed the safety of biosimilar rhGH, demonstrating no marked differences between injection-site reactions induced by lyophilized or liquid formulations of biosimilar rhGH and the reference medicine. All parameters measured in pharmacokinetic and PD studies in healthy volunteers were highly comparable, providing evidence that liquid and lyophilized formulations of biosimilar rhGH were bioequivalent to the reference medicine. Furthermore, data from one pivotal efficacy study (comprising three sub-studies) and a pivotal safety study in children with growth failure secondary to growth hormone deficiency were also submitted. Overall, equivalent therapeutic efficacy and clinical comparability between biosimilar rhGH and the reference medicine were demonstrated. The safety and immunogenicity profiles were comparable with the reference medicine and consistent with clinical experience with rhGH preparations.12
On the basis of this complete package of analytical, nonclinical, and clinical data, the CHMP arrived at a positive opinion for biosimilar rhGH; however, no marketing authorization was granted at that time due to the biosimilar legislation not having been finalized. The sponsor resubmitted its application 2 years later, once Directive 2004/27/EC had come into force, and biosimilar rhGH was eventually approved as the first biosimilar medicine in 2006.
In the USA, meanwhile, a regulatory pathway for the approval of biosimilars was much slower to be enacted. The Patient Protection and Affordable Care Act, signed into law in 2010, created an abbreviated licensure pathway for biosimilars, known as the 351(k) pathway, under the Biologics Price Competition and Innovation Act (BPCIA). Although enacted in 2010, final guidance on the implementation of the 351(k) pathway was not issued until 2015.23 Due to the fact that somatropin was regulated as a drug and not as a biologic, Omnitrope was filed in the USA under the 505(b)(2) approval pathway that allows an applicant to rely on the safety and effectiveness data of a previously approved medicine. However, although the 351(k) pathway relates to products regulated as biologics under the BPCIA, the 505(b)(2) pathway is used for products that are regulated as drugs under the Food Drug and Cosmetic Act.24 Using this pathway (and essentially the same data package that enabled authorization as a biosimilar in the EU), the US Food and Drug Administration (FDA) approved Omnitrope in 2006 as a New Drug Application, not as a biosimilar. In fact, it was 2015 before the first biosimilar (Zarxio® [filgrastim]; Sandoz Inc.) was approved by the US FDA under the 351(k) pathway.25
Although the FDA biosimilar regulatory pathway is generally similar to that of the EMA, some differences do exist. For example, the FDA pathway includes a regulatory designation on interchangeability, which in the USA is the regulatory prerequisite for automatic substitution at the pharmacy level. To be declared interchangeable by the FDA, a biosimilar is expected to have the same clinical result as the reference medicine in any given patient and, if administered more than once to an individual, the risk in terms of safety or diminished efficacy of switching between the biological product and the reference product is not greater than the risk of using the reference medicine alone. The EMA does not have any provision to make assessments of pharmacy-level substitution, rather it is the responsibility of individual states to make this designation. Another difference between the pathways is that the FDA has a requirement for a transition study to grant approval of a biosimilar; in this study, patients who are on the reference medicine are switched to the bio-similar product in development to show that there are no increases in safety events between the pre- and postswitch population. A transition study is not required by the EMA.
Of course, existing guidelines must evolve over time to take account of scientific and technological developments, as well as the accumulated experience with marketing authorization applications and marketed medicinal products. Revised versions of the EMA’s overarching biosimilars guideline and nonclinical and clinical issues, for example, came into effect in 2015. The updated guidance allows clinical trials conducted using reference medicines authorized outside the European Economic Area to be used for the EU filing. In the past, these trials would have had to be repeated in European patients, using an EU-approved reference medicine, at extra cost to the sponsor. To account for the challenges associated with developing more complex biosimilar medicinal medicines, such as biosimilar monoclonal antibodies, several nonclinical and clinical issues were also re-evaluated, including the selection of relevant species for nonclinical studies, need for clinical equivalence studies and other issues regarding the design of the clinical studies, role of biomarkers, amount of immunogenicity data needed, and the possibility to extrapolate to other indications of the reference medicine.
Extrapolation, which is the authorization of the biosimilar for clinical indications of the reference medicine without the need to conduct clinical trials in those indications, is an important element of the biosimilarity concept. Several professional medical societies have discouraged the use of biosimilars in extrapolated indications.26–31 However, from a scientific and regulatory perspective, the active substance of the biosimilar is just another version of the active substance of the originator medicine. As stated previously, the same scientific principles that underlie the comparability exercise for the purpose of demonstrating similarity of a medicine before and after a change in manufacturing process also apply to the comparability exercise for the purpose of demonstrating biosimilarity. When biosimilar comparability has been demonstrated, extrapolation to other indications of the reference medicine could be acceptable, but needs to be scientifically justified and considered in light of all analytical, nonclinical, and clinical data. Additional data would be required if, for example, the active substance of the reference medicine interacts with several receptors that may have a different impact in the tested and nontested therapeutic indications, the active substance itself has more than one active site and the sites may have a different impact in different therapeutic indications, or the studied therapeutic indication is not sensitive enough to detect differences in all relevant aspects of efficacy and safety. The additional data would preferably include PD parameters and/or specific functional assays reflecting the pharmacologic action(s) of the molecule, whereas clinical studies using outcome end points are usually less sensitive for detecting potential differences between the biosimilar and the reference medicine.32
The legal framework for biosimilars enabled the EMA to pioneer the regulatory review of biosimilars according to a new scientific approach, which is also consistently reflected in the product labeling. For reference biologics, all data from the clinical development program are included in the Summary of Product Characteristics (SmPC). For biosimilars, the development program is designed to demonstrate the similarity to the reference medicine on a quality level, complemented by preclinical studies, with clinical data intended only to reconfirm the already demonstrated similarity (and not to reestablish safety and efficacy). Therefore, all new information on the development history of a biosimilar, all nonproprietary product information, and all scientific and regulatory decision-making are found in the European Public Assessment Report of each product, whereas the SmPC of a biosimilar is an essential copy of the reference medicine SmPC and provides the prescriber with all relevant information for safe application and to avoid misunderstanding of the risk/benefit profile of the biosimilar. It is interesting to note that there are no examples of changes to an SmPC label being required for biosimilars that have been approved in Europe; this demonstrates the credibility of the scientific basis and quality of regulatory decision-making by the EMA.
Summary
The EMA has led the way in establishing regulatory pathways for the approval of biosimilars. The credibility of the scientific basis behind the biosimilar concept, and quality of regulatory decision-making, is demonstrated by the successful approval and clinical use of 20 biosimilar medicines since 2006 when biosimilar rhGH was first approved. The regulatory environment for biosimilars continues to evolve, in recognition of advances in technology/analytical methods and the availability of new targets for biosimilar development.
Acknowledgments
Medical writing assistance was provided by Tony Reardon of Spirit Medical Communications Ltd, funded by Sandoz GmbH.
Footnotes
Disclosure
MS and MZ are employees of Sandoz International GmbH/Hexal AG. The authors report no other conflicts of interest in this work.
References
Morrow T, Felcone LH. Defining the difference: what makes biologics unique. Biotechnol Healthc. 2004;1(4):24–29.
Putrik P, Ramiro S, Kvien TK, et al. Working Group “Equity in access to treatment of rheumatoid arthritis in Europe” Inequities in access to biologic and synthetic DMARDs across 46 European countries. Ann Rheum Dis. 2014;73(1):198–206.
Hofmarcher T, Jönsson B, Wilking N. Access to high-quality oncology care across Europe. Lund Swedish Institute for Health Economics; 2014. [Accessed December 8, 2016]. Available from: http://www.ihe.se/access-to-high-quality-oncology.aspx.
Holzmann J, Balser S, Windisch J. Totality of the evidence at work: the first U.S. biosimilar. Expert Opin Biol Ther. 2016;16(2):137–142.
Shaw BE, Confer DL, Hwang WY, Pamphilon DH, Pulsipher MA. Concerns about the use of biosimilar granulocyte colony-stimulating factors for the mobilization of stem cells in normal donors: position of the World Marrow Donor Association. Haematologica. 2011;96(7):942–947.
Barosi G, Bosi A, Abbracchio MP, et al. Key concepts and critical issues on epoetin and filgrastim biosimilars. A position paper from the Italian Society of Hematology, Italian Society of Experimental Hematology, and Italian Group for Bone Marrow Transplantation. Haematologica. 2011;96(7):937–942.
Danese S, Gomollon F, Governing Board and Operational Board of ECCO ECCO position statement: the use of biosimilar medicines in the treatment of inflammatory bowel disease (IBD) J Crohns Colitis. 2013;7(7):586–589.
Fiorino G, Girolomoni G, Lapadula G, et al. The use of biosimilars in immune-mediated disease: a joint Italian Society of Rheumatology (SIR), Italian Society of Dermatology (SIDeMaST), and Italian Group of Inflammatory Bowel Disease (IG-IBD) position paper. Autoimmun Rev. 2014;13(7):751–755.
Fonseca JE, Gonçalves J, Araújo F, et al. Sociedade Portuguesa de Reumatologia The Portuguese Society of Rheumatology position paper on the use of biosimilars. Acta Reumatol Port. 2014;39(1):60–71.
Mularczyk A, Gonciarz M, Bartnik W, et al. Biosimilar medicines – their use in the treatment of inflammatory bowel diseases. Position statement of the Working Group of the Polish National Consultant in Gastroenterology. Prz Gastroenterol. 2014;9(1):1–3.
Weise M, Kurki P, Wolff-Holz E, Bielsky MC, Schneider CK. Biosimilars: the science of extrapolation. Blood. 2014;124(22):3191–3196.

















(2) Pathway: Uncovering Drug Development Trends and Regulatory Requirements")






